Demultiplexing with hashtag oligos (HTOs)
Compiled: 2026-05-22
Source:vignettes/hashing_vignette.Rmd
hashing_vignette.RmdDeveloped in collaboration with the Technology Innovation Group at NYGC, Cell Hashing uses oligo-tagged antibodies against ubiquitously expressed surface proteins to place a “sample barcode” on each single cell, enabling different samples to be multiplexed together and run in a single experiment. For more information, please refer to this paper.
This vignette will give a brief demonstration on how to work with data produced with Cell Hashing in Seurat. Applied to two datasets, we can successfully demultiplex cells to their the original sample-of-origin, and identify cross-sample doublets.
HTODemux() implements the
following procedure:
- We perform a k-medoid clustering on the normalized HTO values, which initially separates cells into K(# of samples)+1 clusters.
- We calculate a ‘negative’ distribution for HTO. For each HTO, we use the cluster with the lowest average value as the negative group.
- For each HTO, we fit a negative binomial distribution to the negative cluster. We use the 0.99 quantile of this distribution as a threshold.
- Based on these thresholds, each cell is classified as positive or negative for each HTO.
- Cells that are positive for more than one HTOs are annotated as doublets.
8-HTO dataset from human PBMCs
- Data represent peripheral blood mononuclear cells (PBMCs) from eight different donors.
- Cells from each donor are uniquely labeled, using CD45 as a hashing antibody.
- Samples were subsequently pooled, and run on a single lane of the the 10X Chromium v2 system.
- You can download the count matrices for RNA and HTO here, or the FASTQ files from GEO
Basic setup
Load packages
Read in data
# Load in the UMI matrix
pbmc.umis <- readRDS("/brahms/shared/vignette-data/pbmc_umi_mtx.rds")
# For generating a hashtag count matrix from FASTQ files, please refer to
# https://github.com/Hoohm/CITE-seq-Count. Load in the HTO count matrix
pbmc.htos <- readRDS("/brahms/shared/vignette-data/pbmc_hto_mtx.rds")
# Select cell barcodes detected by both RNA and HTO In the example datasets we have already
# filtered the cells for you, but perform this step for clarity.
joint.bcs <- intersect(colnames(pbmc.umis), colnames(pbmc.htos))
# Subset RNA and HTO counts by joint cell barcodes
pbmc.umis <- pbmc.umis[, joint.bcs]
pbmc.htos <- as.matrix(pbmc.htos[, joint.bcs])
# Confirm that the HTO have the correct names
rownames(pbmc.htos)## [1] "HTO_A" "HTO_B" "HTO_C" "HTO_D" "HTO_E" "HTO_F" "HTO_G" "HTO_H"Setup Seurat object and add in the HTO data
# Setup Seurat object
pbmc.hashtag <- CreateSeuratObject(counts = Matrix::Matrix(as.matrix(pbmc.umis), sparse = T))
# Normalize RNA data with log normalization
pbmc.hashtag <- NormalizeData(pbmc.hashtag)
# Find and scale variable features
pbmc.hashtag <- FindVariableFeatures(pbmc.hashtag, selection.method = "mean.var.plot")
pbmc.hashtag <- ScaleData(pbmc.hashtag, features = VariableFeatures(pbmc.hashtag))Adding HTO data as an independent assay
You can read more about working with multi-modal data here
# Add HTO data as a new assay independent from RNA
pbmc.hashtag[["HTO"]] <- CreateAssayObject(counts = pbmc.htos)
# Normalize HTO data, here we use centered log-ratio (CLR) transformation
pbmc.hashtag <- NormalizeData(pbmc.hashtag, assay = "HTO", normalization.method = "CLR")Demultiplex cells based on HTO enrichment
Here we use the Seurat function HTODemux() to assign
single cells back to their sample origins.
# If you have a very large dataset we suggest using k_function = 'clara'. This is a k-medoid
# clustering function for large applications You can also play with additional parameters (see
# documentation for HTODemux()) to adjust the threshold for classification Here we are using
# the default settings
pbmc.hashtag <- HTODemux(pbmc.hashtag, assay = "HTO", positive.quantile = 0.99)Visualize demultiplexing results
Output from running HTODemux() is saved in the object
metadata. We can visualize how many cells are classified as singlets,
doublets and negative/ambiguous cells.
# Global classification results
table(pbmc.hashtag$HTO_classification.global)##
## Doublet Negative Singlet
## 2598 346 13972Visualize enrichment for selected HTOs with ridge plots
# Group cells based on the max HTO signal
Idents(pbmc.hashtag) <- "HTO_maxID"
RidgePlot(pbmc.hashtag, assay = "HTO", features = rownames(pbmc.hashtag[["HTO"]])[1:2], ncol = 2)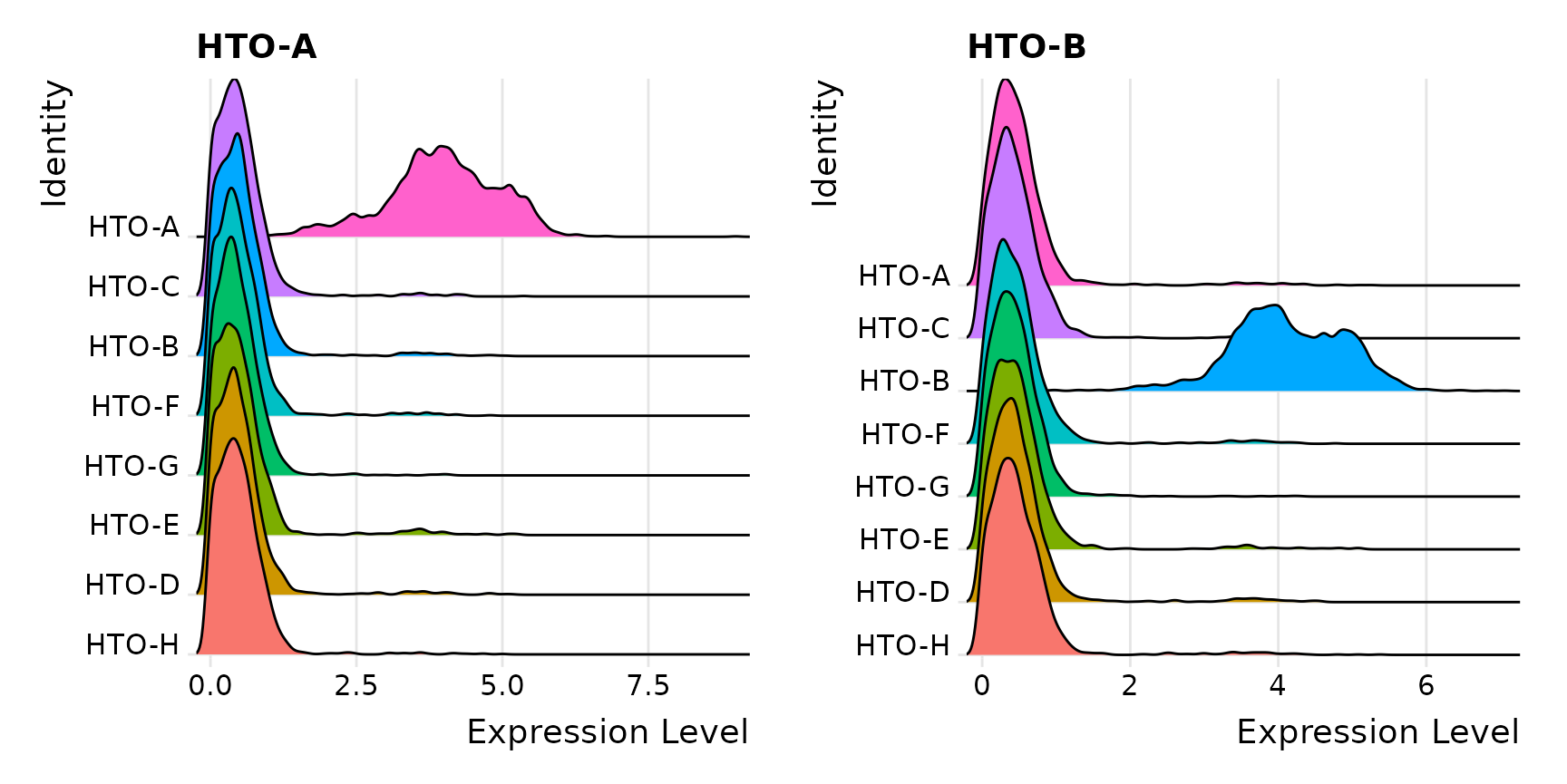
Visualize pairs of HTO signals to confirm mutual exclusivity in singlets
FeatureScatter(pbmc.hashtag, feature1 = "hto_HTO-A", feature2 = "hto_HTO-B")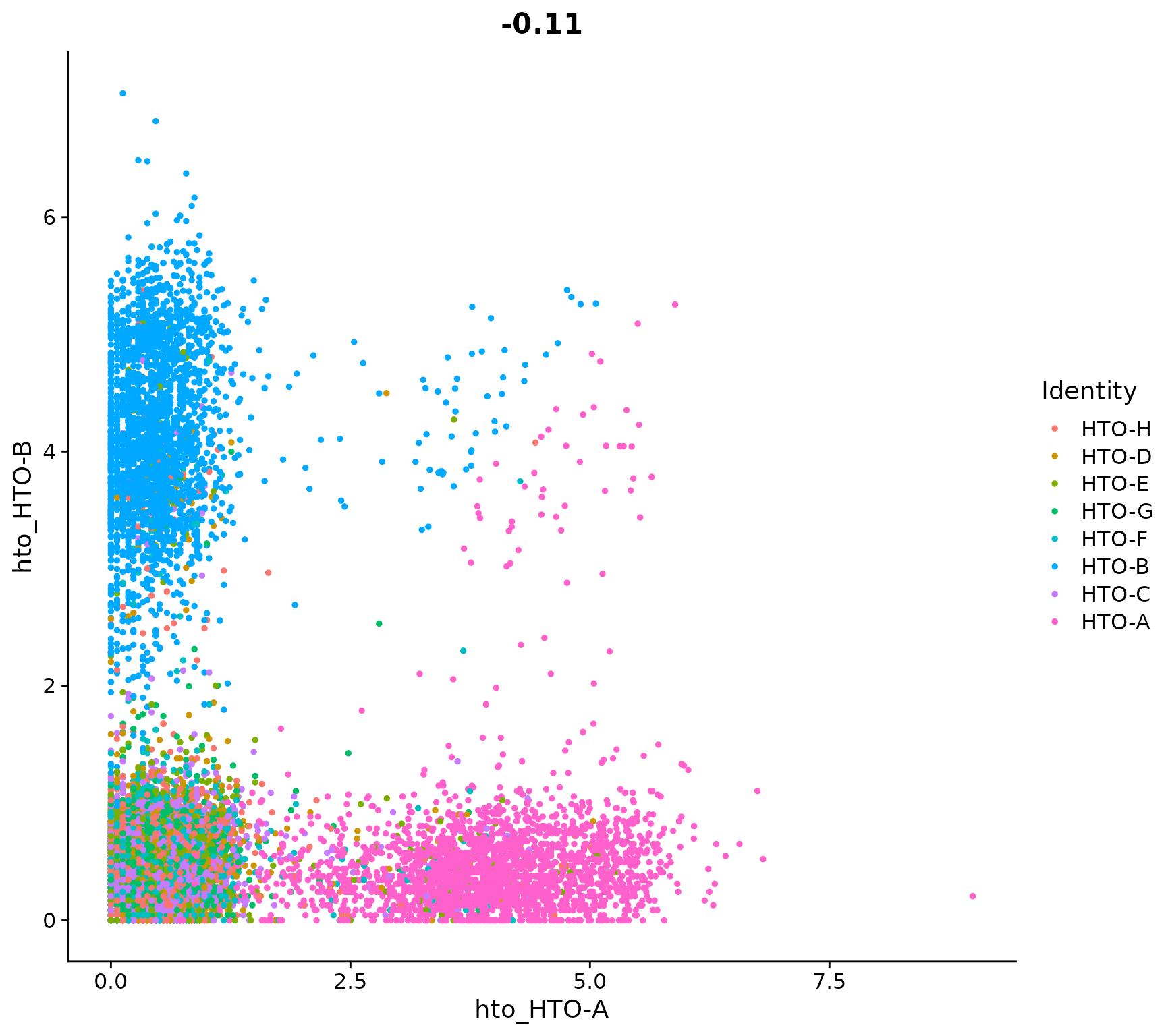
Compare number of UMIs for singlets, doublets and negative cells
Idents(pbmc.hashtag) <- "HTO_classification.global"
VlnPlot(pbmc.hashtag, features = "nCount_RNA", pt.size = 0.1, log = TRUE)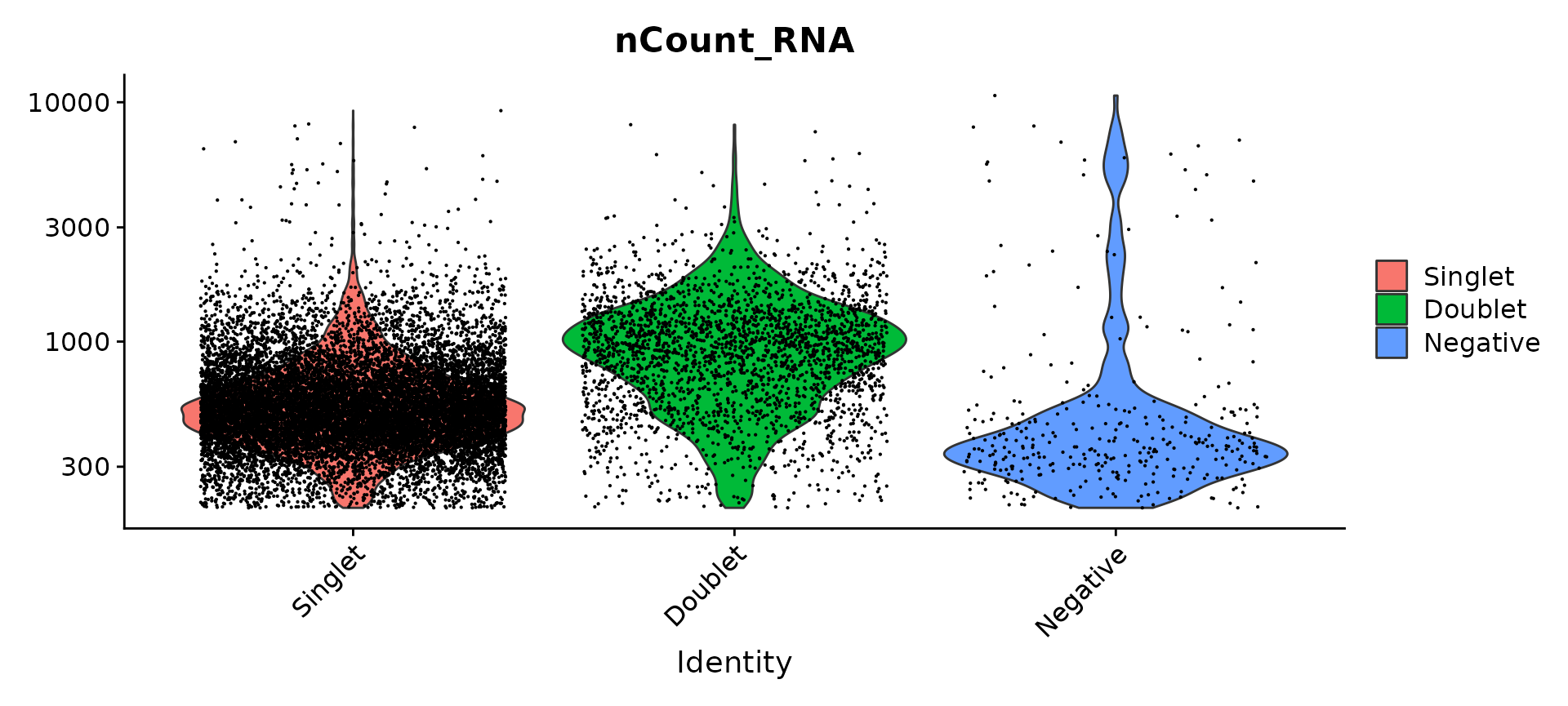
Generate a two dimensional tSNE embedding for HTOs. Here we are grouping cells by singlets and doublets for simplicity.
# First, we will remove negative cells from the object
pbmc.hashtag.subset <- subset(pbmc.hashtag, idents = "Negative", invert = TRUE)
# Calculate a tSNE embedding of the HTO data
DefaultAssay(pbmc.hashtag.subset) <- "HTO"
pbmc.hashtag.subset <- ScaleData(pbmc.hashtag.subset, features = rownames(pbmc.hashtag.subset),
verbose = FALSE)
pbmc.hashtag.subset <- RunPCA(pbmc.hashtag.subset, features = rownames(pbmc.hashtag.subset), approx = FALSE)
pbmc.hashtag.subset <- RunTSNE(pbmc.hashtag.subset, dims = 1:8, perplexity = 100)
DimPlot(pbmc.hashtag.subset)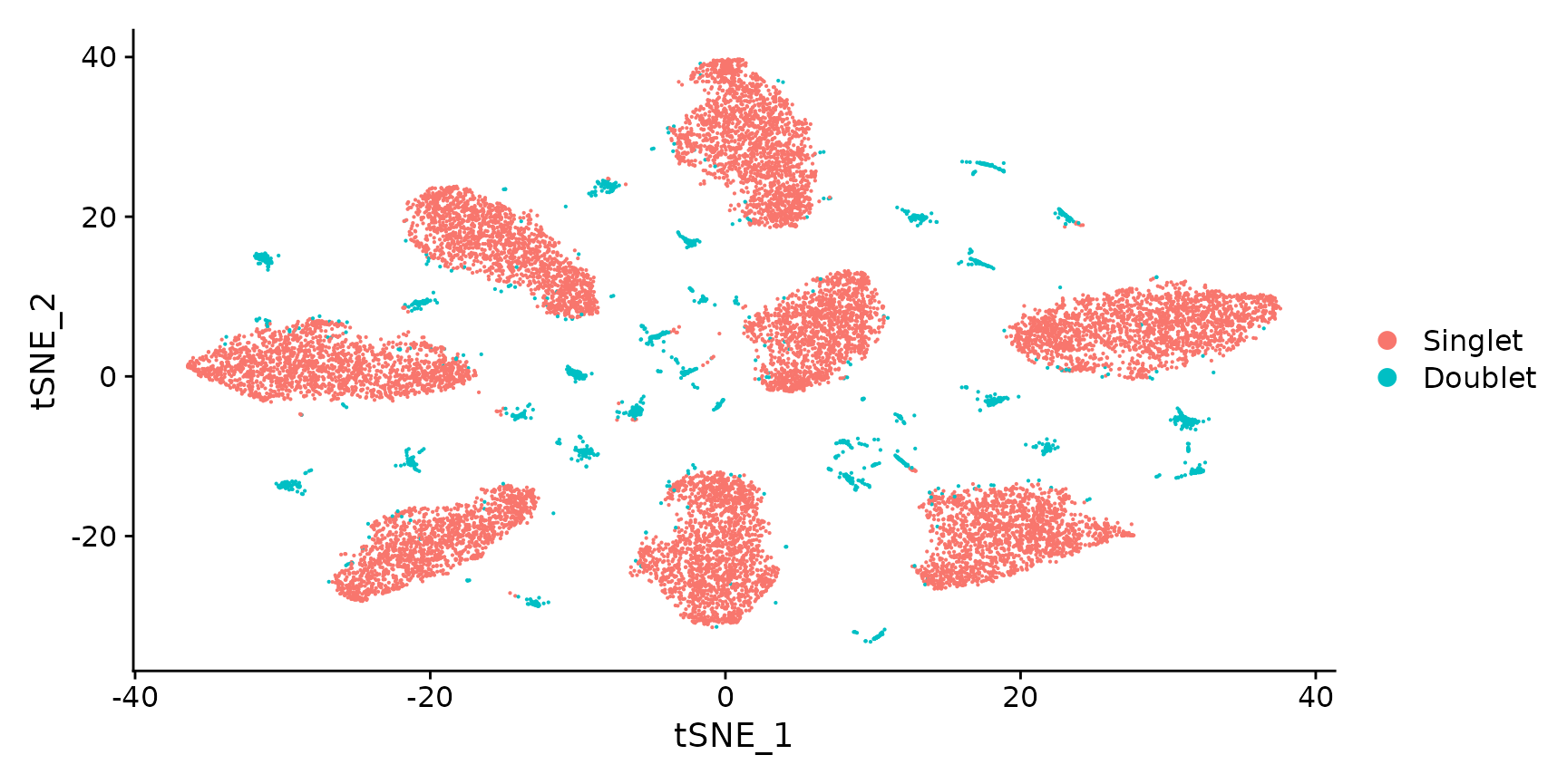
# You can also visualize the more detailed classification result by running Idents(object) <-
# 'HTO_classification' before plotting. Here, you can see that each of the small clouds on the
# tSNE plot corresponds to one of the 28 possible doublet combinations.Create an HTO heatmap, based on Figure 1C in the Cell Hashing paper.
# To increase the efficiency of plotting, you can subsample cells using the num.cells argument
HTOHeatmap(pbmc.hashtag, assay = "HTO", ncells = 5000)
Cluster and visualize cells using the usual scRNA-seq workflow, and examine for the potential presence of batch effects.
# Extract the singlets
pbmc.singlet <- subset(pbmc.hashtag, idents = "Singlet")
# Select the top 1000 most variable features
pbmc.singlet <- FindVariableFeatures(pbmc.singlet, selection.method = "mean.var.plot")
# Scaling RNA data, we only scale the variable features here for efficiency
pbmc.singlet <- ScaleData(pbmc.singlet, features = VariableFeatures(pbmc.singlet))
# Run PCA
pbmc.singlet <- RunPCA(pbmc.singlet, features = VariableFeatures(pbmc.singlet))
# We select the top 10 PCs for clustering and tSNE based on PCElbowPlot
pbmc.singlet <- FindNeighbors(pbmc.singlet, reduction = "pca", dims = 1:10)
pbmc.singlet <- FindClusters(pbmc.singlet, resolution = 0.6, verbose = FALSE)
pbmc.singlet <- RunTSNE(pbmc.singlet, reduction = "pca", dims = 1:10)
# Projecting singlet identities on TSNE visualization
DimPlot(pbmc.singlet, group.by = "HTO_classification")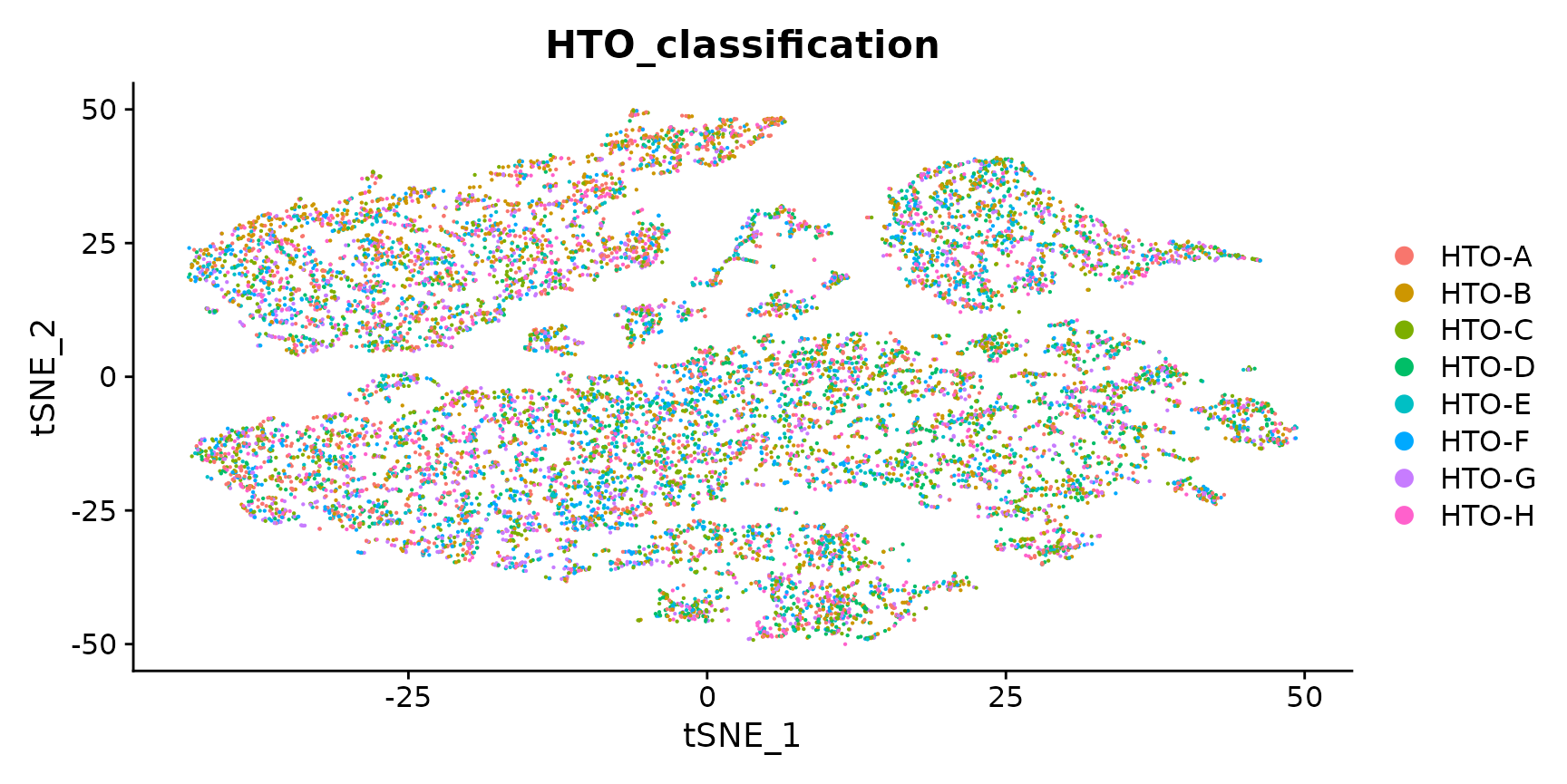
12-HTO dataset from four human cell lines
- Data represent single cells collected from four cell lines: HEK, K562, KG1 and THP1
- Each cell line was further split into three samples (12 samples in total).
- Each sample was labeled with a hashing antibody mixture (CD29 and CD45), pooled, and run on a single lane of 10X.
- Based on this design, we should be able to detect doublets both across and within cell types
- You can download the count matrices for RNA and HTO here, and are available on GEO here
Create Seurat object, add HTO data and perform normalization
# Read in UMI count matrix for RNA
hto12.umis <- readRDS("/brahms/shared/vignette-data/hto12_umi_mtx.rds")
# Read in HTO count matrix
hto12.htos <- readRDS("/brahms/shared/vignette-data/hto12_hto_mtx.rds")
# Select cell barcodes detected in both RNA and HTO
cells.use <- intersect(rownames(hto12.htos), colnames(hto12.umis))
# Create Seurat object and add HTO data
hto12 <- CreateSeuratObject(counts = Matrix::Matrix(as.matrix(hto12.umis[, cells.use]), sparse = T),
min.features = 300)
hto12[["HTO"]] <- CreateAssayObject(counts = t(x = hto12.htos[colnames(hto12), 1:12]))
# Normalize data
hto12 <- NormalizeData(hto12)
hto12 <- NormalizeData(hto12, assay = "HTO", normalization.method = "CLR")Demultiplex data
hto12 <- HTODemux(hto12, assay = "HTO", positive.quantile = 0.99)Visualize demultiplexing results
Distribution of selected HTOs grouped by classification, displayed by ridge plots
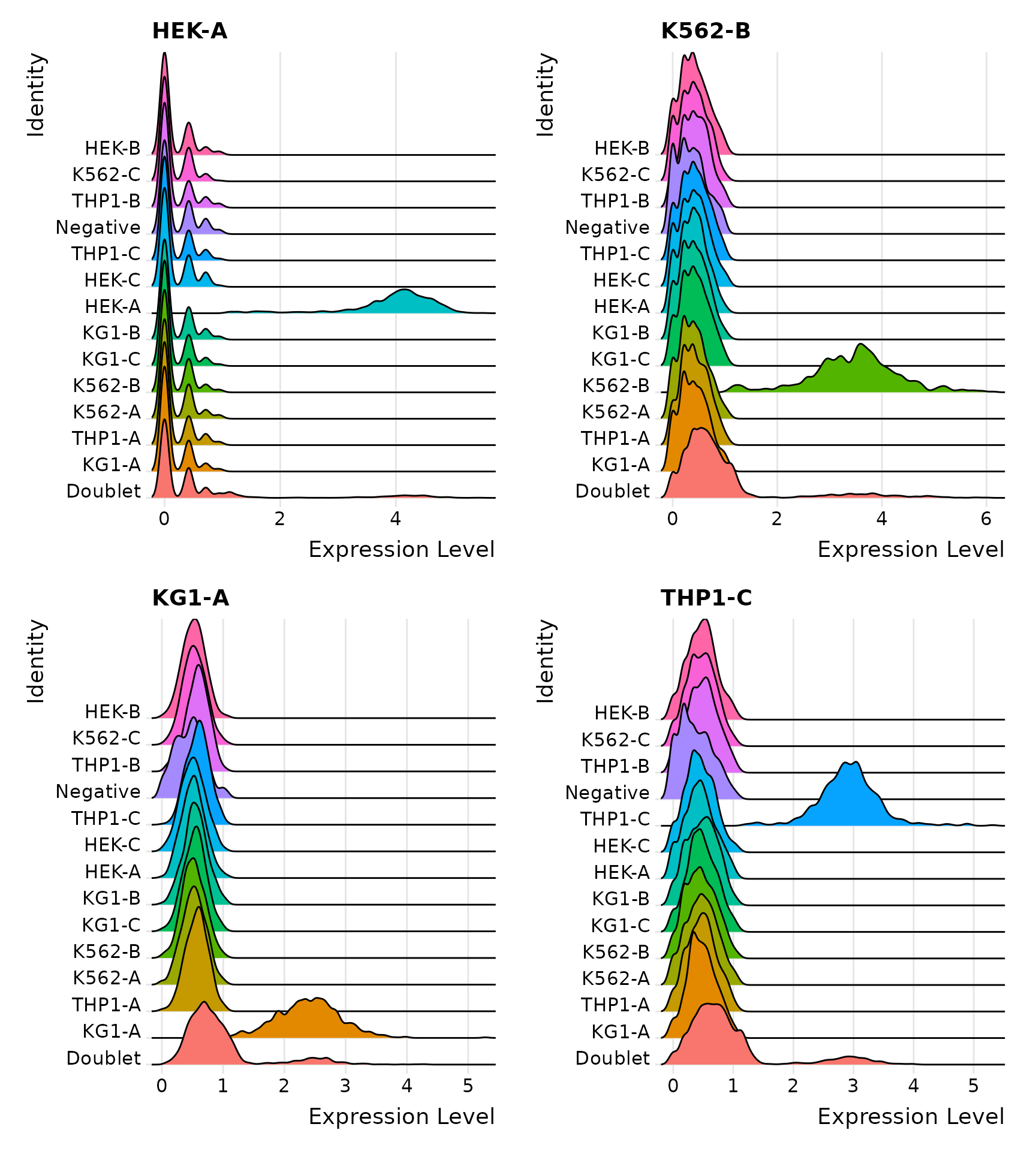
Visualize HTO signals in a heatmap
HTOHeatmap(hto12, assay = "HTO")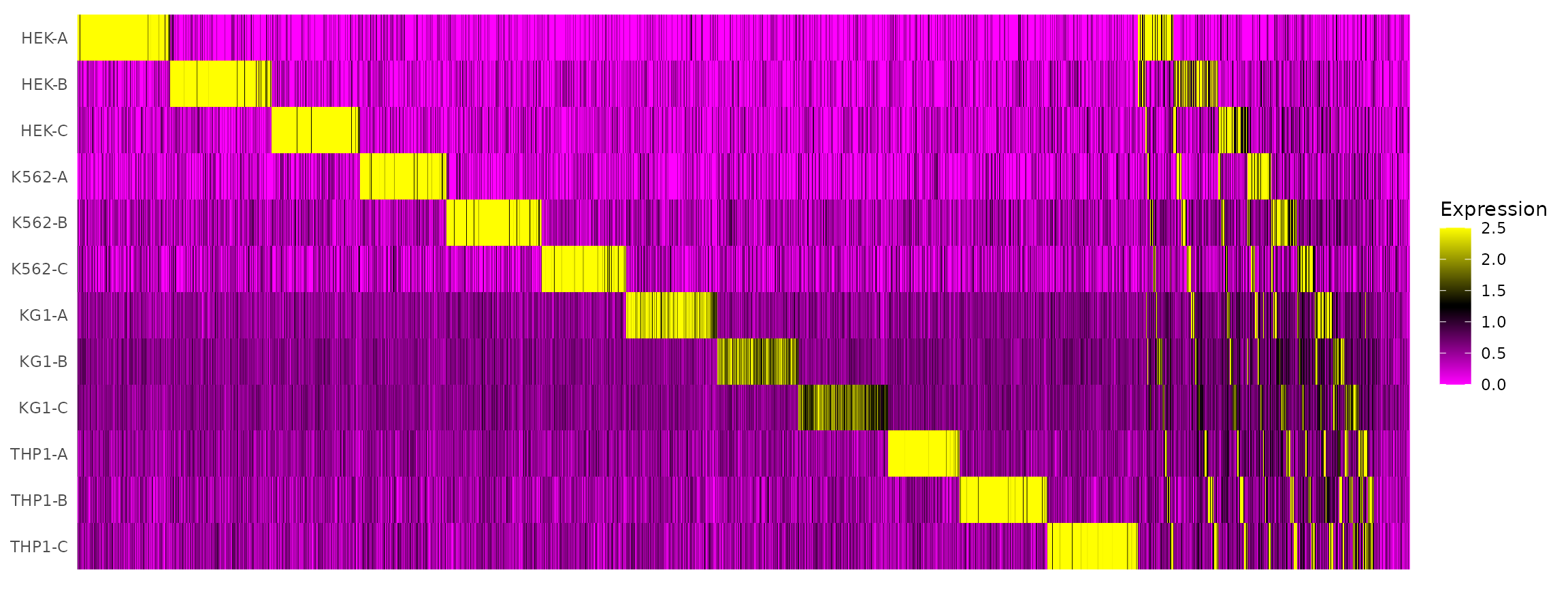
Visualize RNA clustering
# Remove the negative cells
hto12 <- subset(hto12, idents = "Negative", invert = TRUE)
# Run PCA on most variable features
hto12 <- FindVariableFeatures(hto12, selection.method = "mean.var.plot")
hto12 <- ScaleData(hto12, features = VariableFeatures(hto12))
hto12 <- RunPCA(hto12)
hto12 <- RunTSNE(hto12, dims = 1:5, perplexity = 100)
DimPlot(hto12) + NoLegend()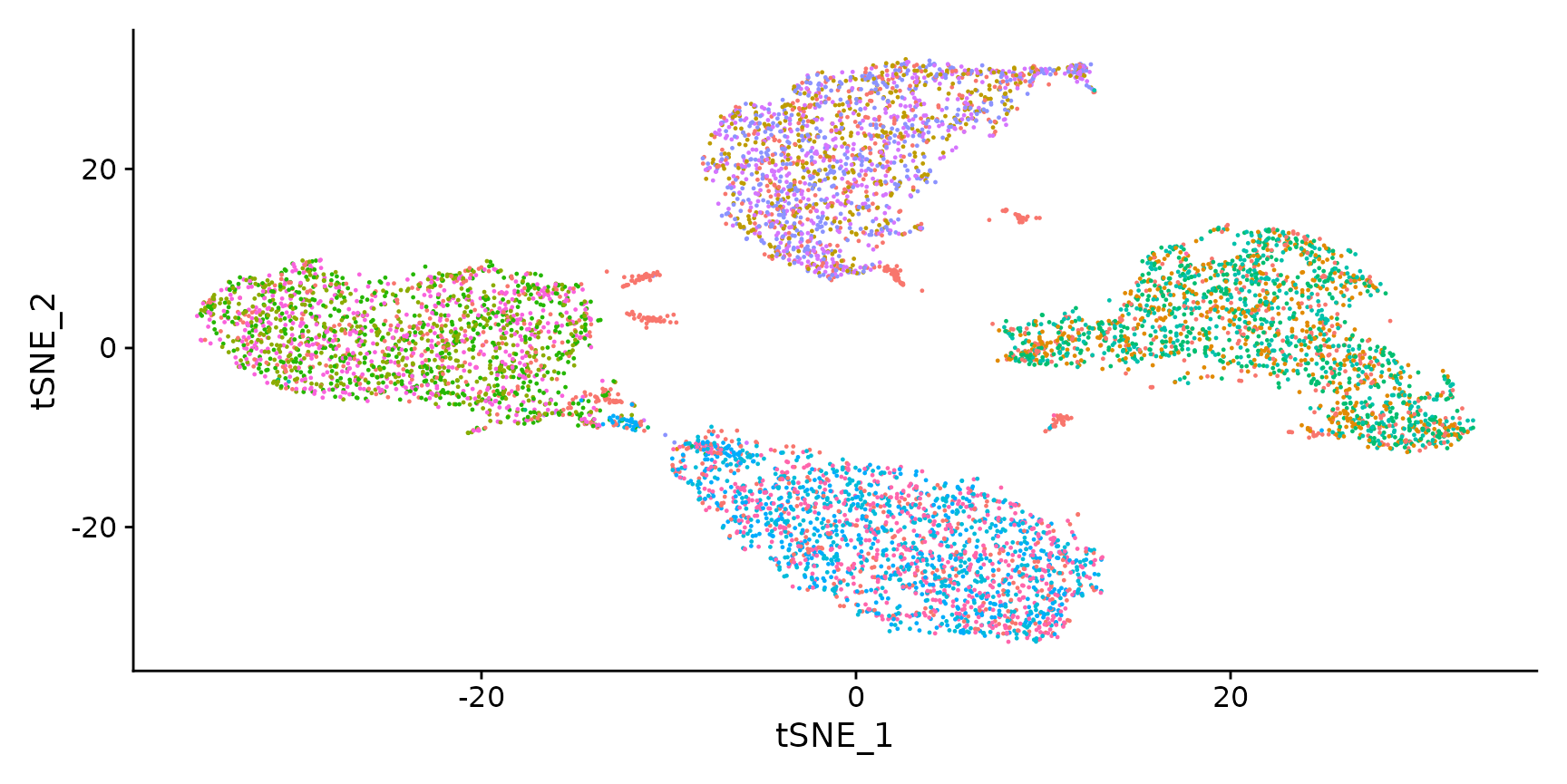
Session Info
## R version 4.5.2 (2025-10-31)
## Platform: x86_64-pc-linux-gnu
## Running under: Ubuntu 24.04.4 LTS
##
## Matrix products: default
## BLAS: /usr/lib/x86_64-linux-gnu/openblas-pthread/libblas.so.3
## LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/libopenblasp-r0.3.26.so; LAPACK version 3.12.0
##
## locale:
## [1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
## [3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
## [5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
## [7] LC_PAPER=en_US.UTF-8 LC_NAME=C
## [9] LC_ADDRESS=C LC_TELEPHONE=C
## [11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
##
## time zone: Etc/UTC
## tzcode source: system (glibc)
##
## attached base packages:
## [1] stats graphics grDevices utils datasets methods base
##
## other attached packages:
## [1] future_1.70.0 Seurat_5.5.0 SeuratObject_5.4.0 sp_2.2-1
##
## loaded via a namespace (and not attached):
## [1] RColorBrewer_1.1-3 jsonlite_2.0.0 magrittr_2.0.5
## [4] spatstat.utils_3.2-2 ggbeeswarm_0.7.3 farver_2.1.2
## [7] rmarkdown_2.30 fs_2.1.0 ragg_1.5.1
## [10] vctrs_0.7.1 ROCR_1.0-12 spatstat.explore_3.8-0
## [13] htmltools_0.5.9 sass_0.4.10 sctransform_0.4.3
## [16] parallelly_1.47.0 KernSmooth_2.23-26 bslib_0.10.0
## [19] htmlwidgets_1.6.4 desc_1.4.3 ica_1.0-3
## [22] plyr_1.8.9 plotly_4.12.0 zoo_1.8-15
## [25] cachem_1.1.0 igraph_2.3.0 mime_0.13
## [28] lifecycle_1.0.5 pkgconfig_2.0.3 Matrix_1.7-5
## [31] R6_2.6.1 fastmap_1.2.0 fitdistrplus_1.2-6
## [34] shiny_1.13.0 digest_0.6.39 patchwork_1.3.2
## [37] tensor_1.5.1 RSpectra_0.16-2 irlba_2.3.7
## [40] textshaping_1.0.5 labeling_0.4.3 progressr_0.19.0
## [43] spatstat.sparse_3.1-0 httr_1.4.8 polyclip_1.10-7
## [46] abind_1.4-8 compiler_4.5.2 withr_3.0.2
## [49] S7_0.2.1 fastDummies_1.7.6 MASS_7.3-65
## [52] tools_4.5.2 vipor_0.4.7 lmtest_0.9-40
## [55] otel_0.2.0 beeswarm_0.4.0 httpuv_1.6.16
## [58] future.apply_1.20.2 goftest_1.2-3 glue_1.8.0
## [61] nlme_3.1-168 promises_1.5.0 grid_4.5.2
## [64] Rtsne_0.17 cluster_2.1.8.2 reshape2_1.4.5
## [67] generics_0.1.4 gtable_0.3.6 spatstat.data_3.1-9
## [70] tidyr_1.3.2 data.table_1.18.2.1 spatstat.geom_3.7-3
## [73] RcppAnnoy_0.0.23 ggrepel_0.9.8 RANN_2.6.2
## [76] pillar_1.11.1 stringr_1.6.0 spam_2.11-3
## [79] RcppHNSW_0.6.0 later_1.4.8 splines_4.5.2
## [82] dplyr_1.2.0 lattice_0.22-7 survival_3.8-3
## [85] deldir_2.0-4 tidyselect_1.2.1 miniUI_0.1.2
## [88] pbapply_1.7-4 knitr_1.51 gridExtra_2.3
## [91] scattermore_1.2 xfun_0.56 matrixStats_1.5.0
## [94] stringi_1.8.7 lazyeval_0.2.3 yaml_2.3.12
## [97] evaluate_1.0.5 codetools_0.2-20 tibble_3.3.1
## [100] cli_3.6.6 uwot_0.2.4 xtable_1.8-8
## [103] reticulate_1.46.0 systemfonts_1.3.2 jquerylib_0.1.4
## [106] Rcpp_1.1.1-1.1 globals_0.19.1 spatstat.random_3.4-5
## [109] png_0.1-9 ggrastr_1.0.2 spatstat.univar_3.1-7
## [112] parallel_4.5.2 pkgdown_2.2.0 ggplot2_4.0.3
## [115] dotCall64_1.2 listenv_0.10.1 viridisLite_0.4.3
## [118] scales_1.4.0 ggridges_0.5.7 purrr_1.2.1
## [121] rlang_1.2.0 cowplot_1.2.0 formatR_1.14