Using sctransform in Seurat
Christoph Hafemeister & Rahul Satija
Compiled: 2023-03-27
Source:vignettes/sctransform_vignette.Rmdsctransform_vignette.RmdBiological heterogeneity in single-cell RNA-seq data is often confounded by technical factors including sequencing depth. The number of molecules detected in each cell can vary significantly between cells, even within the same celltype. Interpretation of scRNA-seq data requires effective pre-processing and normalization to remove this technical variability. In Hafemeister and Satija, 2019 we introduce a modeling framework for the normalization and variance stabilization of molecular count data from scRNA-seq experiment. This procedure omits the need for heuristic steps including pseudocount addition or log-transformation and improves common downstream analytical tasks such as variable gene selection, dimensional reduction, and differential expression.
In this vignette, we demonstrate how using sctransform based normalization enables recovering sharper biological distinction compared to log-normalization.
Load data and create Seurat object
pbmc_data <- Read10X(data.dir = "../data/pbmc3k/filtered_gene_bc_matrices/hg19/")
pbmc <- CreateSeuratObject(counts = pbmc_data)Apply sctransform normalization
- Note that this single command replaces
NormalizeData(),ScaleData(), andFindVariableFeatures(). - Transformed data will be available in the SCT assay, which is set as the default after running sctransform
- During normalization, we can also remove confounding sources of variation, for example, mitochondrial mapping percentage
# store mitochondrial percentage in object meta data
pbmc <- PercentageFeatureSet(pbmc, pattern = "^MT-", col.name = "percent.mt")
# run sctransform
pbmc <- SCTransform(pbmc, vars.to.regress = "percent.mt", verbose = FALSE)The latest version of sctransform also supports using glmGamPoi package which substantially improves the speed of the learning procedure. It can be invoked by specifying method="glmGamPoi".
if (!requireNamespace("BiocManager", quietly = TRUE)) install.packages("BiocManager")
BiocManager::install("glmGamPoi")
pbmc <- SCTransform(pbmc, method = "glmGamPoi", vars.to.regress = "percent.mt", verbose = FALSE)Perform dimensionality reduction by PCA and UMAP embedding
# These are now standard steps in the Seurat workflow for visualization and clustering
pbmc <- RunPCA(pbmc, verbose = FALSE)
pbmc <- RunUMAP(pbmc, dims = 1:30, verbose = FALSE)
pbmc <- FindNeighbors(pbmc, dims = 1:30, verbose = FALSE)
pbmc <- FindClusters(pbmc, verbose = FALSE)
DimPlot(pbmc, label = TRUE) + NoLegend()In the standard Seurat workflow we focus on 10 PCs for this dataset, though we highlight that the results are similar with higher settings for this parameter. Interestingly, we’ve found that when using sctransform, we often benefit by pushing this parameter even higher. We believe this is because the sctransform workflow performs more effective normalization, strongly removing technical effects from the data. Even after standard log-normalization, variation in sequencing depth is still a confounding factor (see Figure 1), and this effect can subtly influence higher PCs. In sctransform, this effect is substantially mitigated (see Figure 3). This means that higher PCs are more likely to represent subtle, but biologically relevant, sources of heterogeneity – so including them may improve downstream analysis. In addition, sctransform returns 3,000 variable features by default, instead of 2,000. The rationale is similar, the additional variable features are less likely to be driven by technical differences across cells, and instead may represent more subtle biological fluctuations. In general, we find that results produced with sctransform are less dependent on these parameters (indeed, we achieve nearly identical results when using all genes in the transcriptome, though this does reduce computational efficiency). This can help users generate more robust results, and in addition, enables the application of standard analysis pipelines with identical parameter settings that can quickly be applied to new datasets: For example, the following code replicates the full end-to-end workflow, in a single command: As described in our paper, sctransform calculates a model of technical noise in scRNA-seq data using ‘regularized negative binomial regression’. The residuals for this model are normalized values, and can be positive or negative. Positive residuals for a given gene in a given cell indicate that we observed more UMIs than expected given the gene’s average expression in the population and cellular sequencing depth, while negative residuals indicate the converse. The results of sctransfrom are stored in the “SCT” assay. You can learn more about multi-assay data and commands in Seurat in our vignette, command cheat sheet, or developer guide.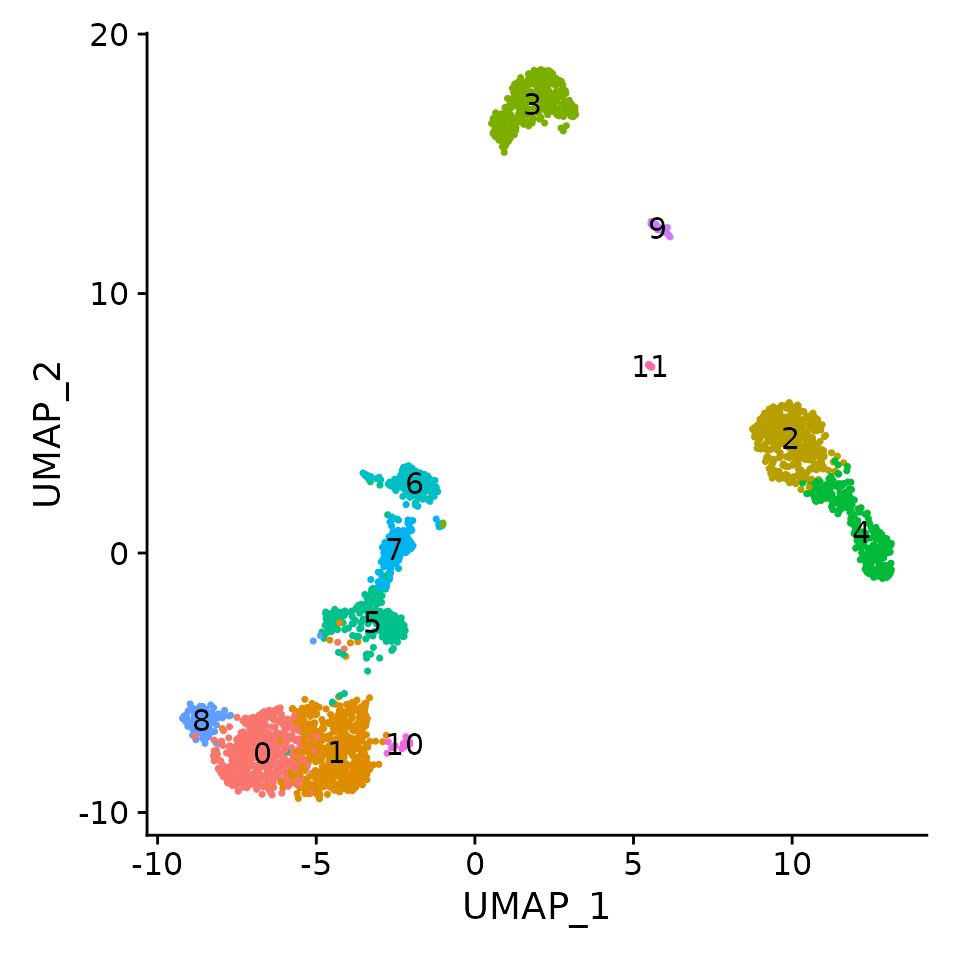
Why can we choose more PCs when using sctransform?
pbmc <- CreateSeuratObject(pbmc_data) %>%
PercentageFeatureSet(pattern = "^MT-", col.name = "percent.mt") %>%
SCTransform(vars.to.regress = "percent.mt") %>%
RunPCA() %>%
FindNeighbors(dims = 1:30) %>%
RunUMAP(dims = 1:30) %>%
FindClusters()Where are normalized values stored for sctransform?
pbmc[["SCT"]]@scale.data contains the residuals (normalized values), and is used directly as input to PCA. Please note that this matrix is non-sparse, and can therefore take up a lot of memory if stored for all genes. To save memory, we store these values only for variable genes, by setting the return.only.var.genes = TRUE by default in the SCTransform() function call.pbmc[["SCT"]]@counts. We store log-normalized versions of these corrected counts in pbmc[["SCT"]]@data, which are very helpful for visualization.scale.data slot) themselves. This is not currently supported in Seurat v3, but will be soon.
Users can individually annotate clusters based on canonical markers. However, the sctransform normalization reveals sharper biological distinctions compared to the standard Seurat workflow, in a few ways:
- Clear separation of at least 3 CD8 T cell populations (naive, memory, effector), based on CD8A, GZMK, CCL5, GZMK expression
- Clear separation of three CD4 T cell populations (naive, memory, IFN-activated) based on S100A4, CCR7, IL32, and ISG15
- Additional developmental sub-structure in B cell cluster, based on TCL1A, FCER2
- Additional separation of NK cells into CD56dim vs. bright clusters, based on XCL1 and FCGR3A
# These are now standard steps in the Seurat workflow for visualization and clustering
# Visualize canonical marker genes as violin plots.
VlnPlot(pbmc, features = c("CD8A", "GZMK", "CCL5", "S100A4", "ANXA1", "CCR7", "ISG15", "CD3D"),
pt.size = 0.2, ncol = 4)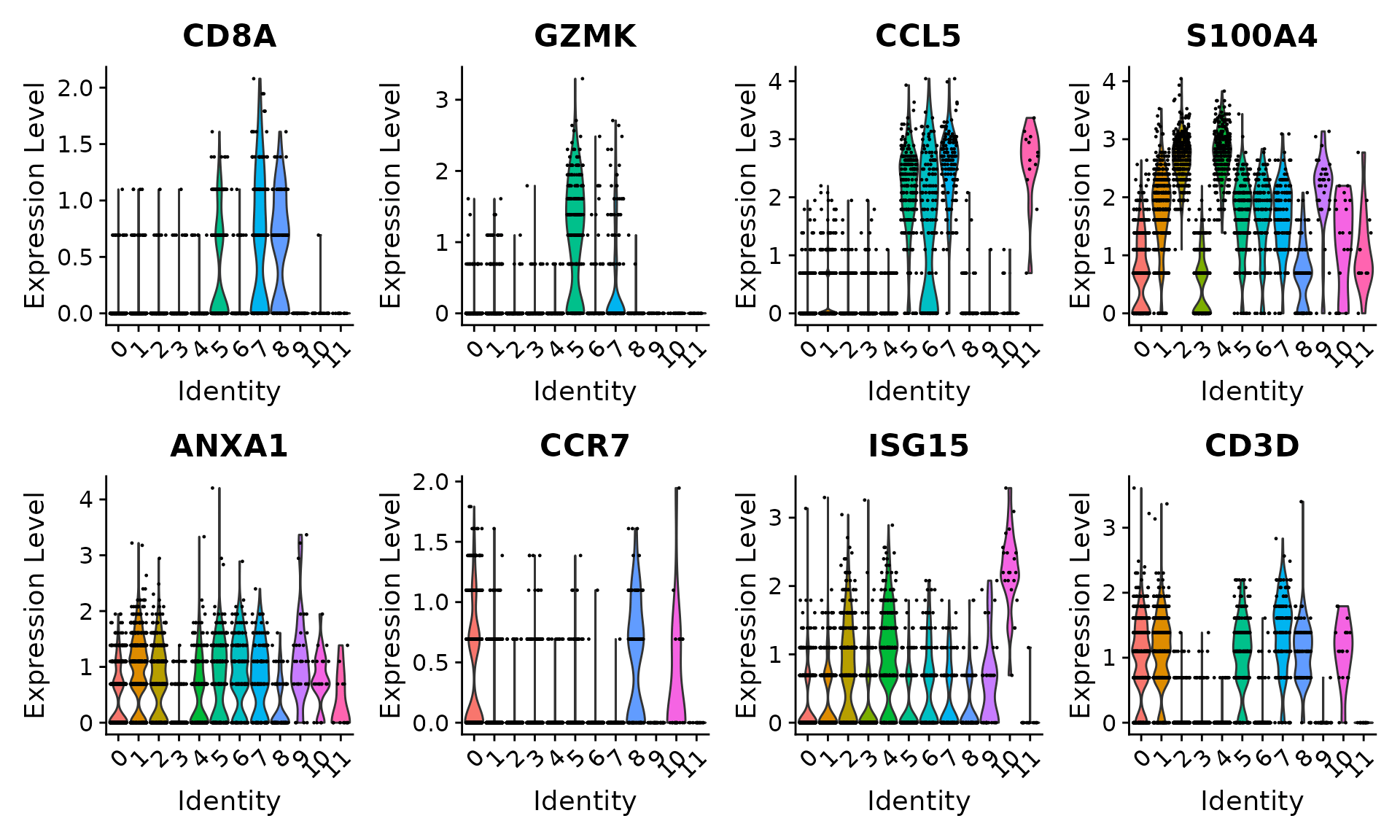
# Visualize canonical marker genes on the sctransform embedding.
FeaturePlot(pbmc, features = c("CD8A", "GZMK", "CCL5", "S100A4", "ANXA1", "CCR7"), pt.size = 0.2,
ncol = 3)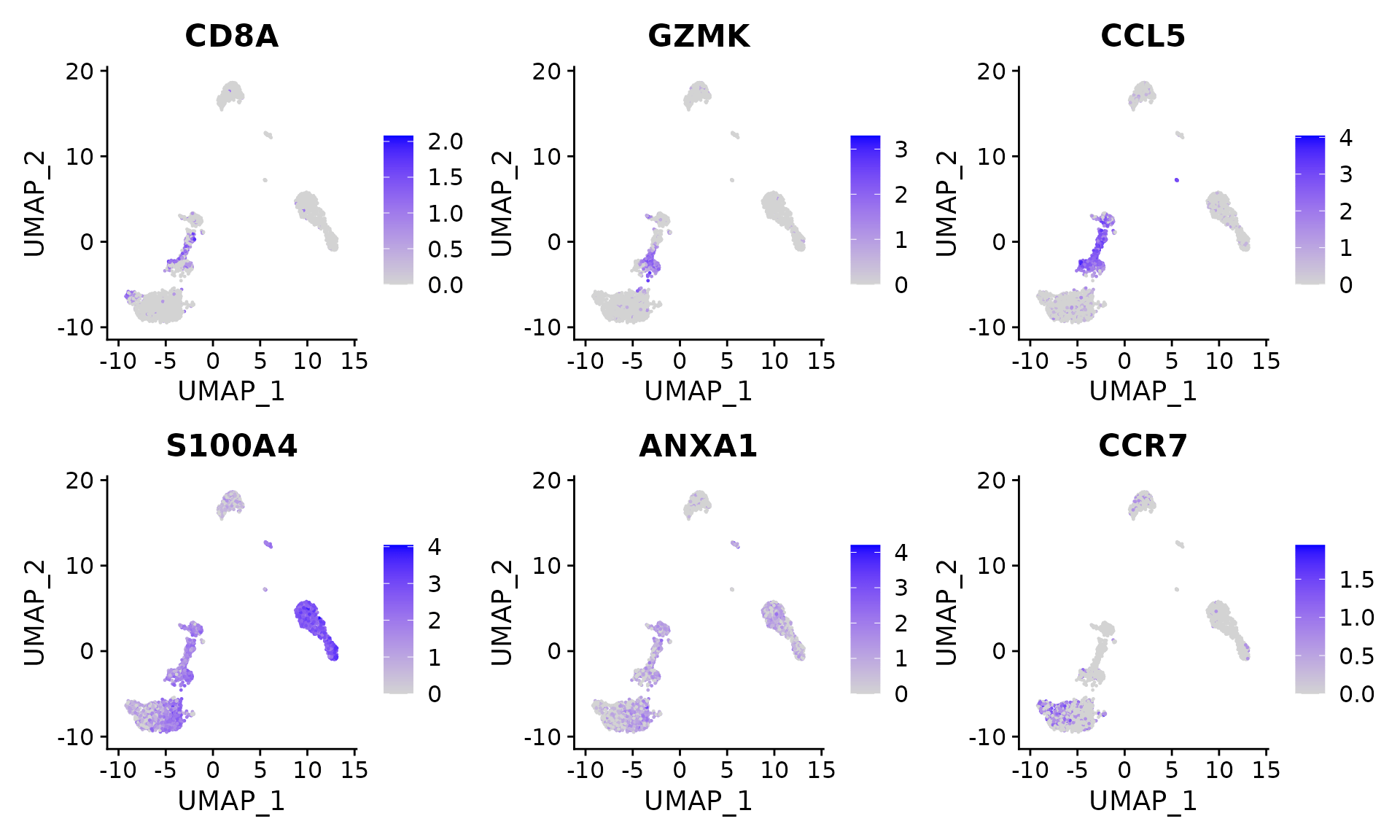
FeaturePlot(pbmc, features = c("CD3D", "ISG15", "TCL1A", "FCER2", "XCL1", "FCGR3A"), pt.size = 0.2,
ncol = 3)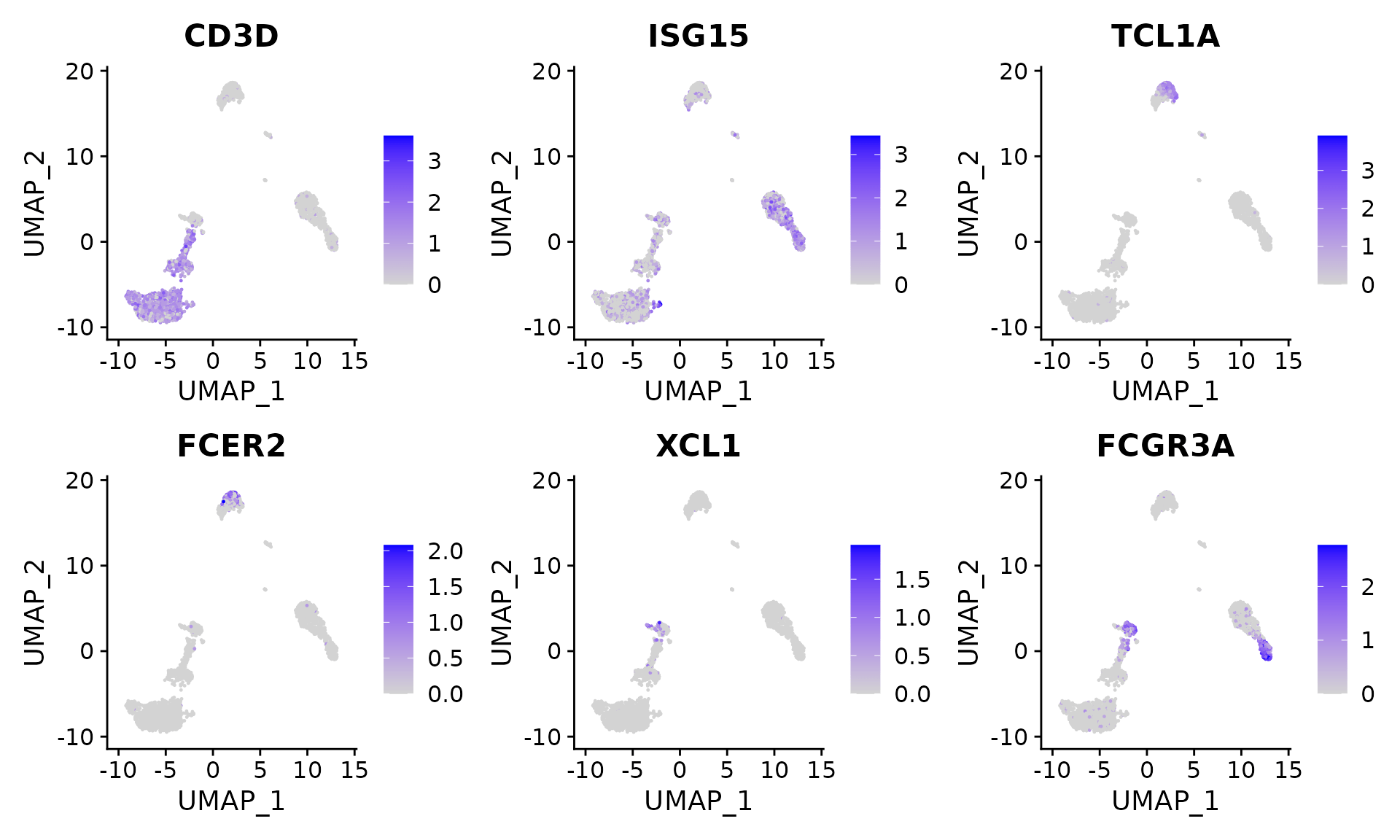
Session Info
## R version 4.2.0 (2022-04-22)
## Platform: x86_64-pc-linux-gnu (64-bit)
## Running under: Ubuntu 20.04.5 LTS
##
## Matrix products: default
## BLAS: /usr/lib/x86_64-linux-gnu/openblas-pthread/libblas.so.3
## LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/liblapack.so.3
##
## locale:
## [1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
## [3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
## [5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
## [7] LC_PAPER=en_US.UTF-8 LC_NAME=C
## [9] LC_ADDRESS=C LC_TELEPHONE=C
## [11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
##
## attached base packages:
## [1] stats graphics grDevices utils datasets methods base
##
## other attached packages:
## [1] sctransform_0.3.5 ggplot2_3.4.1 SeuratObject_4.1.3 Seurat_4.3.0
##
## loaded via a namespace (and not attached):
## [1] Rtsne_0.16 colorspace_2.1-0 deldir_1.0-6
## [4] ellipsis_0.3.2 ggridges_0.5.4 rprojroot_2.0.3
## [7] fs_1.6.1 spatstat.data_3.0-0 farver_2.1.1
## [10] leiden_0.4.3 listenv_0.9.0 ggrepel_0.9.3
## [13] fansi_1.0.4 R.methodsS3_1.8.2 codetools_0.2-18
## [16] splines_4.2.0 cachem_1.0.7 knitr_1.42
## [19] polyclip_1.10-4 jsonlite_1.8.4 ica_1.0-3
## [22] cluster_2.1.3 R.oo_1.25.0 png_0.1-8
## [25] uwot_0.1.14 spatstat.sparse_3.0-0 shiny_1.7.4
## [28] compiler_4.2.0 httr_1.4.5 Matrix_1.5-3
## [31] fastmap_1.1.1 lazyeval_0.2.2 cli_3.6.0
## [34] later_1.3.0 formatR_1.14 htmltools_0.5.4
## [37] tools_4.2.0 igraph_1.4.1 gtable_0.3.1
## [40] glue_1.6.2 RANN_2.6.1 reshape2_1.4.4
## [43] dplyr_1.1.0 Rcpp_1.0.10 scattermore_0.8
## [46] jquerylib_0.1.4 pkgdown_2.0.7 vctrs_0.5.2
## [49] nlme_3.1-157 spatstat.explore_3.0-6 progressr_0.13.0
## [52] lmtest_0.9-40 spatstat.random_3.1-3 xfun_0.37
## [55] stringr_1.5.0 globals_0.16.2 mime_0.12
## [58] miniUI_0.1.1.1 lifecycle_1.0.3 irlba_2.3.5.1
## [61] goftest_1.2-3 future_1.31.0 MASS_7.3-56
## [64] zoo_1.8-11 scales_1.2.1 ragg_1.2.5
## [67] promises_1.2.0.1 spatstat.utils_3.0-1 parallel_4.2.0
## [70] RColorBrewer_1.1-3 yaml_2.3.7 memoise_2.0.1
## [73] reticulate_1.28 pbapply_1.7-0 gridExtra_2.3
## [76] sass_0.4.5 stringi_1.7.12 highr_0.10
## [79] desc_1.4.2 rlang_1.0.6 pkgconfig_2.0.3
## [82] systemfonts_1.0.4 matrixStats_0.63.0 evaluate_0.20
## [85] lattice_0.20-45 tensor_1.5 ROCR_1.0-11
## [88] purrr_1.0.1 labeling_0.4.2 patchwork_1.1.2
## [91] htmlwidgets_1.6.1 cowplot_1.1.1 tidyselect_1.2.0
## [94] parallelly_1.34.0 RcppAnnoy_0.0.20 plyr_1.8.8
## [97] magrittr_2.0.3 R6_2.5.1 generics_0.1.3
## [100] withr_2.5.0 pillar_1.8.1 fitdistrplus_1.1-8
## [103] abind_1.4-5 survival_3.3-1 sp_1.6-0
## [106] tibble_3.1.8 future.apply_1.10.0 KernSmooth_2.23-20
## [109] utf8_1.2.3 spatstat.geom_3.0-6 plotly_4.10.1
## [112] rmarkdown_2.20 grid_4.2.0 data.table_1.14.8
## [115] digest_0.6.31 xtable_1.8-4 tidyr_1.3.0
## [118] httpuv_1.6.9 R.utils_2.12.2 textshaping_0.3.6
## [121] munsell_0.5.0 viridisLite_0.4.1 bslib_0.4.2