Seurat - Interaction Tips
Compiled: March 27, 2023
Source:vignettes/interaction_vignette.Rmdinteraction_vignette.RmdLoad in the data
This vignette demonstrates some useful features for interacting with the Seurat object. For demonstration purposes, we will be using the 2,700 PBMC object that is created in the first guided tutorial. You can load the data from our SeuratData package. To simulate the scenario where we have two replicates, we will randomly assign half the cells in each cluster to be from “rep1” and other half from “rep2”.
library(Seurat)
library(SeuratData)
InstallData("pbmc3k")
pbmc <- LoadData("pbmc3k", type = "pbmc3k.final")
# pretend that cells were originally assigned to one of two replicates (we assign randomly
# here) if your cells do belong to multiple replicates, and you want to add this info to the
# Seurat object create a data frame with this information (similar to replicate.info below)
set.seed(42)
pbmc$replicate <- sample(c("rep1", "rep2"), size = ncol(pbmc), replace = TRUE)Switch identity class between cluster ID and replicate
# Plot UMAP, coloring cells by cell type (currently stored in object@ident)
DimPlot(pbmc, reduction = "umap")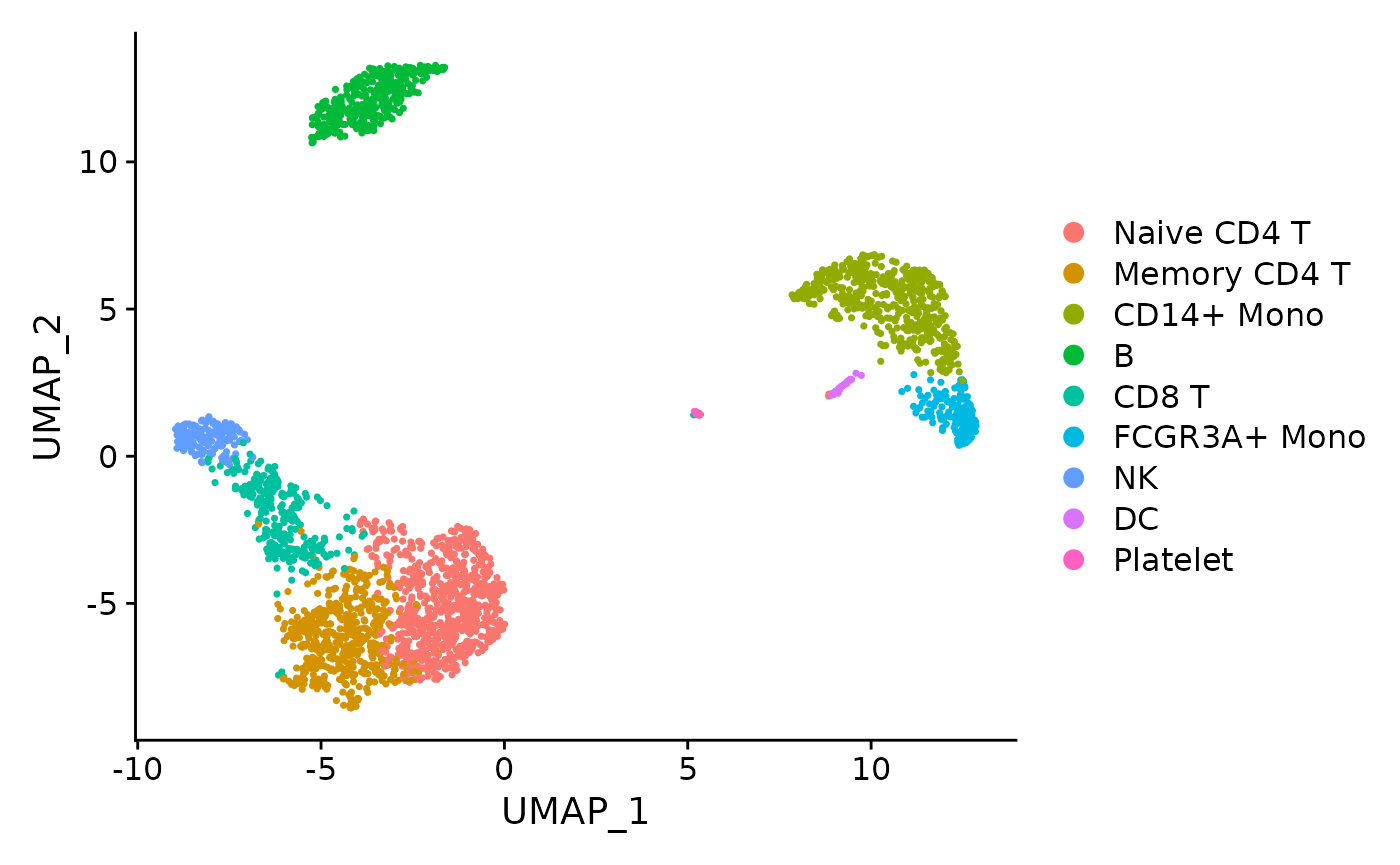
# How do I create a UMAP plot where cells are colored by replicate? First, store the current
# identities in a new column of meta.data called CellType
pbmc$CellType <- Idents(pbmc)
# Next, switch the identity class of all cells to reflect replicate ID
Idents(pbmc) <- "replicate"
DimPlot(pbmc, reduction = "umap")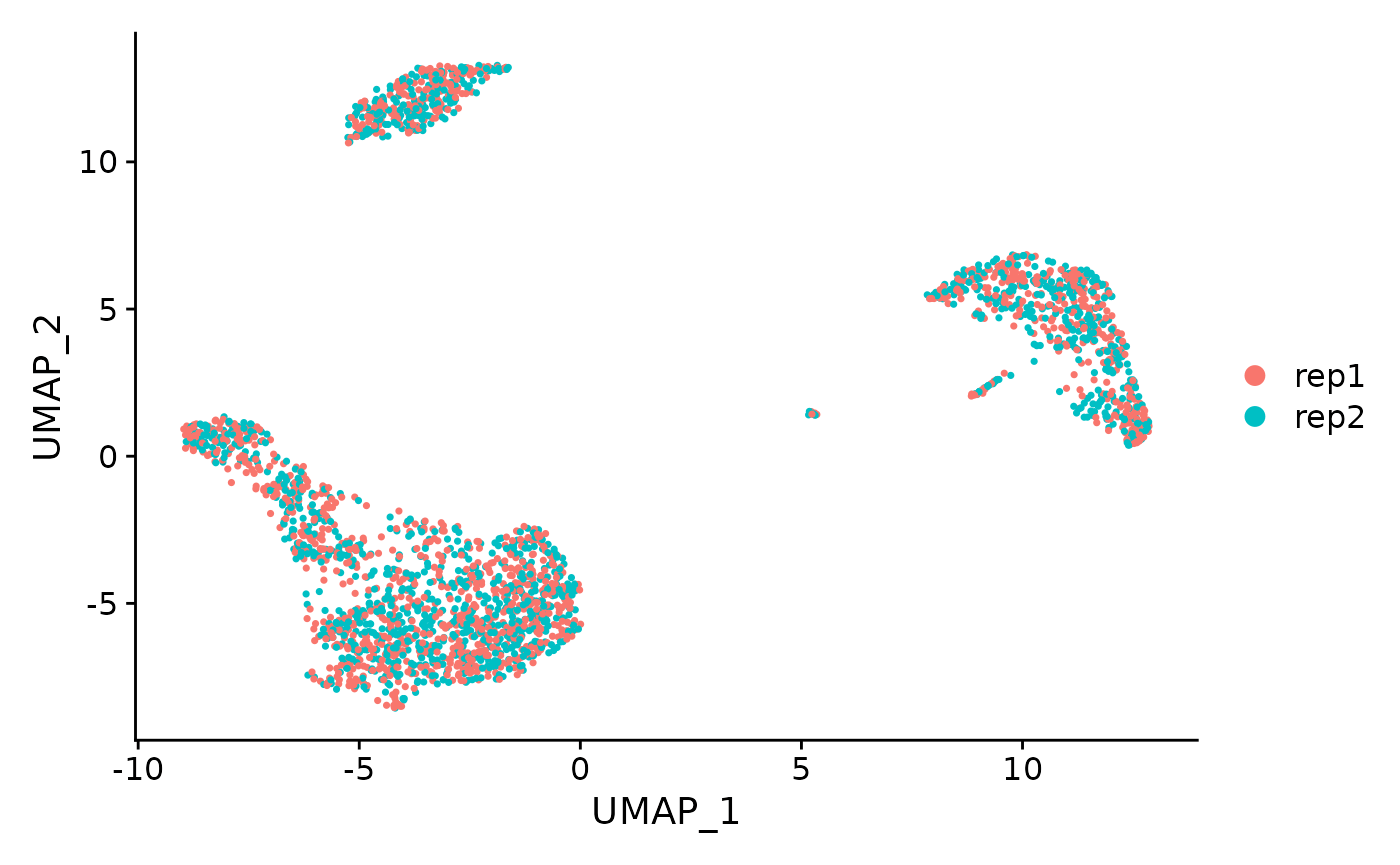
# alternately : DimPlot(pbmc, reduction = 'umap', group.by = 'replicate') you can pass the
# shape.by to label points by both replicate and cell type
# Switch back to cell type labels
Idents(pbmc) <- "CellType"Tabulate cells by cluster ID, replicate, or both
##
## Naive CD4 T Memory CD4 T CD14+ Mono B CD8 T FCGR3A+ Mono
## 697 483 480 344 271 162
## NK DC Platelet
## 155 32 14
# How many cells are in each replicate?
table(pbmc$replicate)##
## rep1 rep2
## 1348 1290
# What proportion of cells are in each cluster?
prop.table(table(Idents(pbmc)))##
## Naive CD4 T Memory CD4 T CD14+ Mono B CD8 T FCGR3A+ Mono
## 0.264215315 0.183093252 0.181956027 0.130401820 0.102729340 0.061410159
## NK DC Platelet
## 0.058756634 0.012130402 0.005307051##
## rep1 rep2
## Naive CD4 T 354 343
## Memory CD4 T 249 234
## CD14+ Mono 232 248
## B 173 171
## CD8 T 154 117
## FCGR3A+ Mono 81 81
## NK 81 74
## DC 18 14
## Platelet 6 8
prop.table(table(Idents(pbmc), pbmc$replicate), margin = 2)##
## rep1 rep2
## Naive CD4 T 0.262611276 0.265891473
## Memory CD4 T 0.184718101 0.181395349
## CD14+ Mono 0.172106825 0.192248062
## B 0.128338279 0.132558140
## CD8 T 0.114243323 0.090697674
## FCGR3A+ Mono 0.060089021 0.062790698
## NK 0.060089021 0.057364341
## DC 0.013353116 0.010852713
## Platelet 0.004451039 0.006201550Selecting particular cells and subsetting the Seurat object
# What are the cell names of all NK cells?
WhichCells(pbmc, idents = "NK")## [1] "AAACCGTGTATGCG" "AAATTCGATTCTCA" "AACCTTACGCGAGA" "AACGCCCTCGTACA"
## [5] "AACGTCGAGTATCG" "AAGATTACCTCAAG" "AAGCAAGAGCTTAG" "AAGCAAGAGGTGTT"
## [9] "AAGTAGGATACAGC" "AATACTGAATTGGC" "AATCCTTGGTGAGG" "AATCTCTGCTTTAC"
## [13] "ACAAATTGTTGCGA" "ACAACCGAGGGATG" "ACAATTGATGACTG" "ACACCCTGGTGTTG"
## [17] "ACAGGTACTGGTGT" "ACCTGGCTAAGTAG" "ACGAACACCTTGTT" "ACGATCGAGGACTT"
## [21] "ACGCAATGGTTCAG" "ACGCTGCTGTTCTT" "ACGGAACTCAGATC" "ACGTGATGTGACAC"
## [25] "ACGTTGGAGCCAAT" "ACTGCCACTCCGTC" "ACTGGCCTTCAGTG" "ACTTCAACGTAGGG"
## [29] "AGAACAGAAATGCC" "AGATATACCCGTAA" "AGATTCCTGTTCAG" "AGCCTCTGCCAATG"
## [33] "AGCGATTGAGATCC" "AGGATGCTTTAGGC" "AGGGACGAGTCAAC" "AGTAATACATCACG"
## [37] "AGTCACGATGAGCT" "AGTTTGCTACTGGT" "ATACCACTGCCAAT" "ATACTCTGGTATGC"
## [41] "ATCCCGTGCAGTCA" "ATCTTTCTTGTCCC" "ATGAAGGACTTGCC" "ATGATAACTTCACT"
## [45] "ATGATATGGTGCTA" "ATGGACACGCATCA" "ATGGGTACATCGGT" "ATTAACGATGAGAA"
## [49] "ATTCCAACTTAGGC" "CAAGGTTGTCTGGA" "CAATCTACTGACTG" "CACCACTGGCGAAG"
## [53] "CACGGGTGGAGGAC" "CAGATGACATTCTC" "CAGCAATGGAGGGT" "CAGCGGACCTTTAC"
## [57] "CAGCTCTGTGTGGT" "CAGTTTACACACGT" "CATCAGGACTTCCG" "CATCAGGATAGCCA"
## [61] "CATGAGACGTTGAC" "CATTACACCAACTG" "CATTTCGAGATACC" "CCTCGAACACTTTC"
## [65] "CGACCACTAAAGTG" "CGACCACTGCCAAT" "CGAGGCTGACGCTA" "CGCCGAGAGCTTAG"
## [69] "CGGCGAACGACAAA" "CGGCGAACTACTTC" "CGGGCATGTCTCTA" "CGTACCTGGCATCA"
## [73] "CGTGTAGACGATAC" "CGTGTAGAGTTACG" "CGTGTAGATTCGGA" "CTAAACCTCTGACA"
## [77] "CTAACGGAACCGAT" "CTACGCACTGGTCA" "CTACTCCTATGTCG" "CTAGTTACGAAACA"
## [81] "CTATACTGCTACGA" "CTATACTGTCTCAT" "CTCGACTGGTTGAC" "CTGAGAACGTAAAG"
## [85] "CTTTAGTGACGGGA" "GAACCAACTTCCGC" "GAAGTGCTAAACGA" "GAATGCACCTTCGC"
## [89] "GAATTAACGTCGTA" "GACGGCACACGGGA" "GAGCGCTGAAGATG" "GAGGTACTGACACT"
## [93] "GAGGTGGATCCTCG" "GATAGAGAAGGGTG" "GATCCCTGACCTTT" "GCACACCTGTGCTA"
## [97] "GCACCACTTCCTTA" "GCACTAGAGTCGTA" "GCAGGGCTATCGAC" "GCCGGAACGTTCTT"
## [101] "GCCTACACAGTTCG" "GCGCATCTTGCTCC" "GCGCGATGGTGCAT" "GGAAGGTGGCGAGA"
## [105] "GGACGCTGTCCTCG" "GGAGGCCTCGTTGA" "GGCAAGGAAAAAGC" "GGCATATGCTTATC"
## [109] "GGCCGAACTCTAGG" "GGCTAAACACCTGA" "GGGTTAACGTGCAT" "GGTGGAGAAACGGG"
## [113] "GTAGTGTGAGCGGA" "GTCGACCTGAATGA" "GTGATTCTGGTTCA" "GTGTATCTAGTAGA"
## [117] "GTTAAAACCGAGAG" "GTTCAACTGGGACA" "GTTGACGATATCGG" "TAACTCACTCTACT"
## [121] "TAAGAGGACTTGTT" "TAATGCCTCGTCTC" "TACGGCCTGGGACA" "TACTACTGATGTCG"
## [125] "TACTCTGAATCGAC" "TACTGTTGAGGCGA" "TAGCATCTCAGCTA" "TAGCCCACAGCTAC"
## [129] "TAGGGACTGAACTC" "TAGTGGTGAAGTGA" "TAGTTAGAACCACA" "TATGAATGGAGGAC"
## [133] "TATGGGTGCATCAG" "TATTTCCTGGAGGT" "TCAACACTGTTTGG" "TCAGACGACGTTAG"
## [137] "TCCCGAACACAGTC" "TCCTAAACCGCATA" "TCGATTTGCAGCTA" "TCTAACACCAGTTG"
## [141] "TGATAAACTCCGTC" "TGCACAGACGACAT" "TGCCACTGCGATAC" "TGCTGAGAGAGCAG"
## [145] "TGGAACACAAACAG" "TGGTAGACCCTCAC" "TGTAATGACACAAC" "TGTAATGAGGTAAA"
## [149] "TTACTCGATCTACT" "TTAGTCTGCCAACA" "TTCCAAACTCCCAC" "TTCCCACTTGAGGG"
## [153] "TTCTAGTGGAGAGC" "TTCTGATGGAGACG" "TTGTCATGGACGGA"
# How can I extract expression matrix for all NK cells (perhaps, to load into another package)
nk.raw.data <- as.matrix(GetAssayData(pbmc, slot = "counts")[, WhichCells(pbmc, ident = "NK")])
# Can I create a Seurat object based on expression of a feature or value in object metadata?
subset(pbmc, subset = MS4A1 > 1)## An object of class Seurat
## 13714 features across 414 samples within 1 assay
## Active assay: RNA (13714 features, 2000 variable features)
## 2 dimensional reductions calculated: pca, umap
subset(pbmc, subset = replicate == "rep2")## An object of class Seurat
## 13714 features across 1290 samples within 1 assay
## Active assay: RNA (13714 features, 2000 variable features)
## 2 dimensional reductions calculated: pca, umap
# Can I create a Seurat object of just the NK cells and B cells?
subset(pbmc, idents = c("NK", "B"))## An object of class Seurat
## 13714 features across 499 samples within 1 assay
## Active assay: RNA (13714 features, 2000 variable features)
## 2 dimensional reductions calculated: pca, umap
# Can I create a Seurat object of all cells except the NK cells and B cells?
subset(pbmc, idents = c("NK", "B"), invert = TRUE)## An object of class Seurat
## 13714 features across 2139 samples within 1 assay
## Active assay: RNA (13714 features, 2000 variable features)
## 2 dimensional reductions calculated: pca, umap
# note that if you wish to perform additional rounds of clustering after subsetting we
# recommend re-running FindVariableFeatures() and ScaleData()Calculating the average gene expression within a cluster
# How can I calculate the average expression of all cells within a cluster?
cluster.averages <- AverageExpression(pbmc)
head(cluster.averages[["RNA"]][, 1:5])## Naive CD4 T Memory CD4 T CD14+ Mono B CD8 T
## AL627309.1 0.006128664 0.005927264 0.04854338 0.00000000 0.02054586
## AP006222.2 0.000000000 0.008206078 0.01088471 0.00000000 0.01191488
## RP11-206L10.2 0.007453092 0.000000000 0.00000000 0.02065031 0.00000000
## RP11-206L10.9 0.000000000 0.000000000 0.01050116 0.00000000 0.00000000
## LINC00115 0.019118933 0.024690483 0.03753737 0.03888541 0.01948277
## NOC2L 0.497463190 0.359811462 0.27253750 0.58653489 0.55704897
# Return this information as a Seurat object (enables downstream plotting and analysis) First,
# replace spaces with underscores '_' so ggplot2 doesn't fail
orig.levels <- levels(pbmc)
Idents(pbmc) <- gsub(pattern = " ", replacement = "_", x = Idents(pbmc))
orig.levels <- gsub(pattern = " ", replacement = "_", x = orig.levels)
levels(pbmc) <- orig.levels
cluster.averages <- AverageExpression(pbmc, return.seurat = TRUE)
cluster.averages## An object of class Seurat
## 13714 features across 9 samples within 1 assay
## Active assay: RNA (13714 features, 0 variable features)
# How can I plot the average expression of NK cells vs. CD8 T cells? Pass do.hover = T for an
# interactive plot to identify gene outliers
CellScatter(cluster.averages, cell1 = "NK", cell2 = "CD8_T")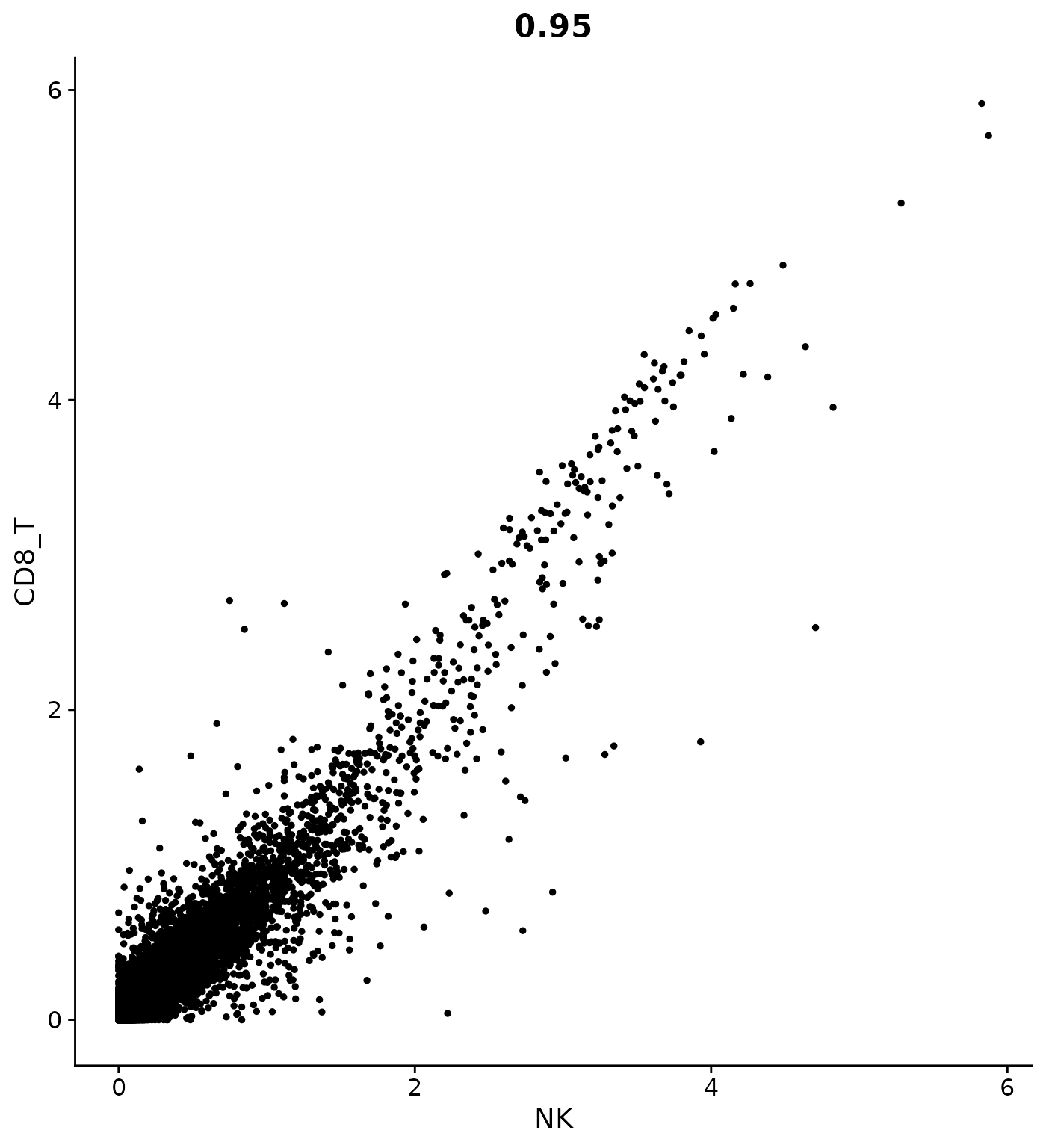
# How can I calculate expression averages separately for each replicate?
cluster.averages <- AverageExpression(pbmc, return.seurat = TRUE, add.ident = "replicate")
CellScatter(cluster.averages, cell1 = "CD8_T_rep1", cell2 = "CD8_T_rep2")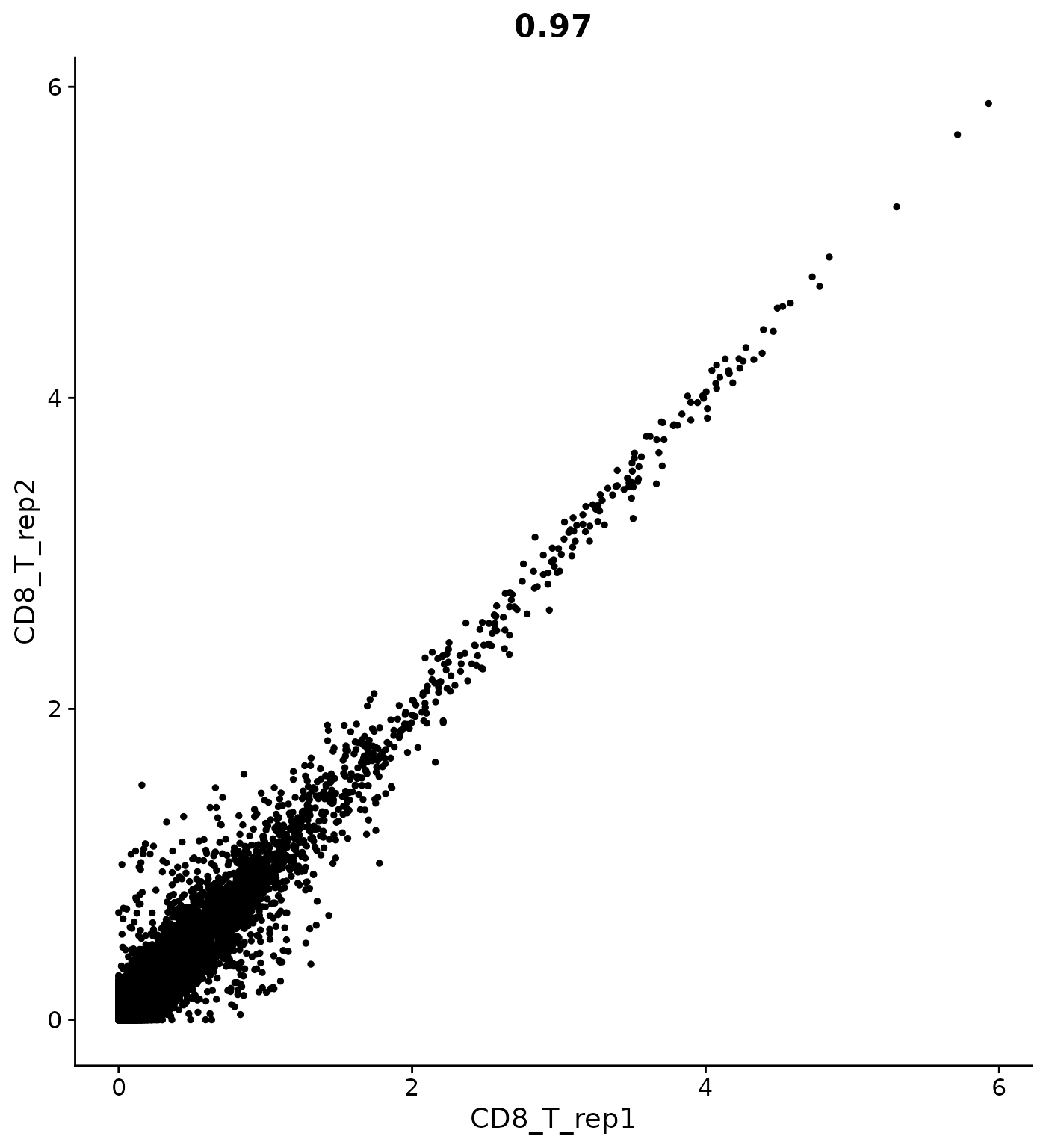
# You can also plot heatmaps of these 'in silico' bulk datasets to visualize agreement between
# replicates
DoHeatmap(cluster.averages, features = unlist(TopFeatures(pbmc[["pca"]], balanced = TRUE)), size = 3,
draw.lines = FALSE)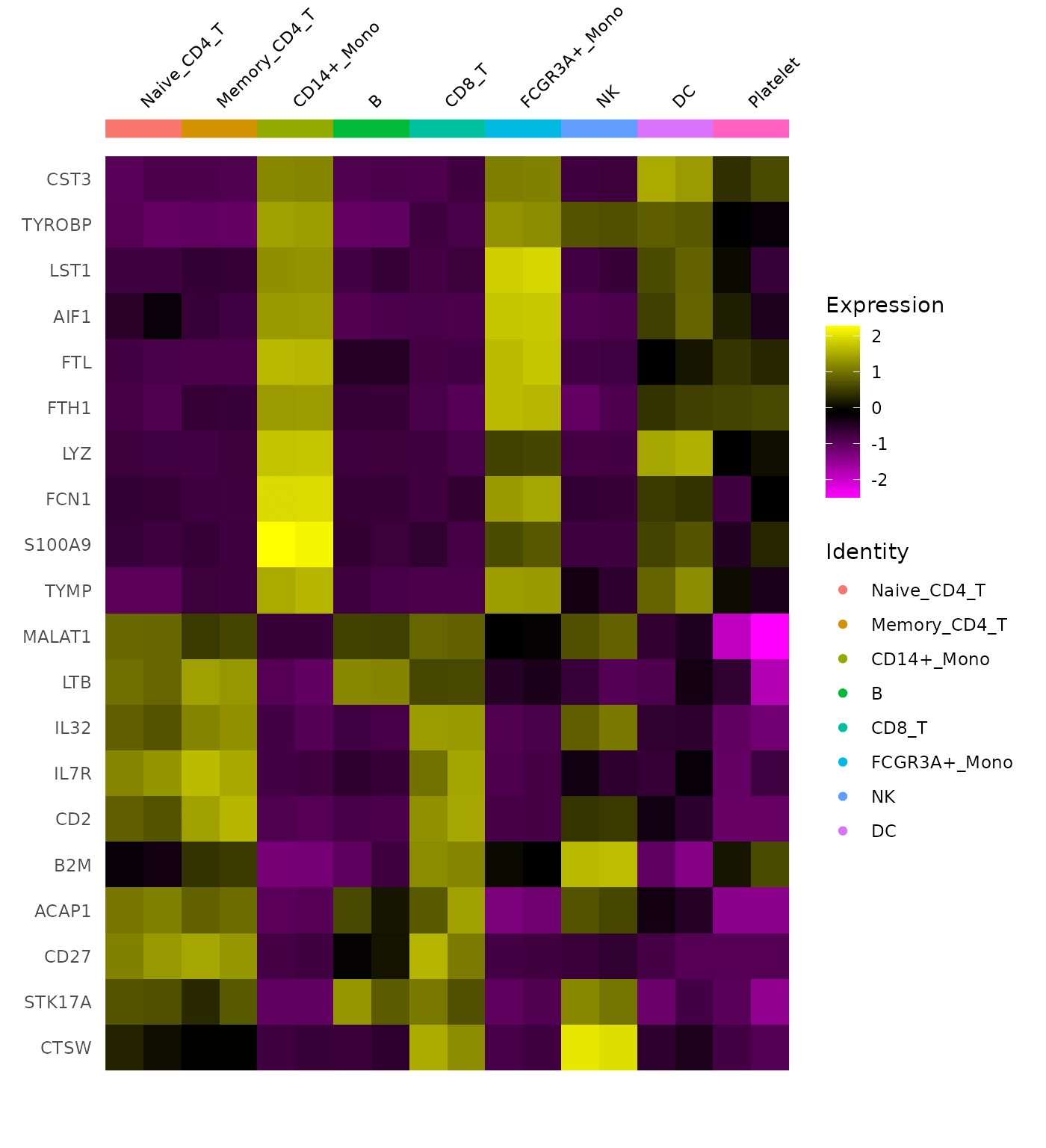
Session Info
## R version 4.2.0 (2022-04-22)
## Platform: x86_64-pc-linux-gnu (64-bit)
## Running under: Ubuntu 20.04.5 LTS
##
## Matrix products: default
## BLAS: /usr/lib/x86_64-linux-gnu/openblas-pthread/libblas.so.3
## LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/liblapack.so.3
##
## locale:
## [1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
## [3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
## [5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
## [7] LC_PAPER=en_US.UTF-8 LC_NAME=C
## [9] LC_ADDRESS=C LC_TELEPHONE=C
## [11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
##
## attached base packages:
## [1] stats graphics grDevices utils datasets methods base
##
## other attached packages:
## [1] thp1.eccite.SeuratData_3.1.5 stxBrain.SeuratData_0.1.1
## [3] ssHippo.SeuratData_3.1.4 pbmcsca.SeuratData_3.0.0
## [5] pbmcMultiome.SeuratData_0.1.2 pbmc3k.SeuratData_3.1.4
## [7] panc8.SeuratData_3.0.2 ifnb.SeuratData_3.1.0
## [9] hcabm40k.SeuratData_3.0.0 bmcite.SeuratData_0.3.0
## [11] SeuratData_0.2.2 SeuratObject_4.1.3
## [13] Seurat_4.3.0
##
## loaded via a namespace (and not attached):
## [1] systemfonts_1.0.4 plyr_1.8.8 igraph_1.4.1
## [4] lazyeval_0.2.2 sp_1.6-0 splines_4.2.0
## [7] listenv_0.9.0 scattermore_0.8 ggplot2_3.4.1
## [10] digest_0.6.31 htmltools_0.5.4 fansi_1.0.4
## [13] magrittr_2.0.3 memoise_2.0.1 tensor_1.5
## [16] cluster_2.1.3 ROCR_1.0-11 globals_0.16.2
## [19] matrixStats_0.63.0 pkgdown_2.0.7 spatstat.sparse_3.0-0
## [22] colorspace_2.1-0 rappdirs_0.3.3 ggrepel_0.9.3
## [25] textshaping_0.3.6 xfun_0.37 dplyr_1.1.0
## [28] crayon_1.5.2 jsonlite_1.8.4 progressr_0.13.0
## [31] spatstat.data_3.0-0 survival_3.3-1 zoo_1.8-11
## [34] glue_1.6.2 polyclip_1.10-4 gtable_0.3.1
## [37] leiden_0.4.3 future.apply_1.10.0 abind_1.4-5
## [40] scales_1.2.1 spatstat.random_3.1-3 miniUI_0.1.1.1
## [43] Rcpp_1.0.10 viridisLite_0.4.1 xtable_1.8-4
## [46] reticulate_1.28 htmlwidgets_1.6.1 httr_1.4.5
## [49] RColorBrewer_1.1-3 ellipsis_0.3.2 ica_1.0-3
## [52] farver_2.1.1 pkgconfig_2.0.3 sass_0.4.5
## [55] uwot_0.1.14 deldir_1.0-6 utf8_1.2.3
## [58] tidyselect_1.2.0 labeling_0.4.2 rlang_1.0.6
## [61] reshape2_1.4.4 later_1.3.0 munsell_0.5.0
## [64] tools_4.2.0 cachem_1.0.7 cli_3.6.0
## [67] generics_0.1.3 ggridges_0.5.4 evaluate_0.20
## [70] stringr_1.5.0 fastmap_1.1.1 yaml_2.3.7
## [73] ragg_1.2.5 goftest_1.2-3 knitr_1.42
## [76] fs_1.6.1 fitdistrplus_1.1-8 purrr_1.0.1
## [79] RANN_2.6.1 pbapply_1.7-0 future_1.31.0
## [82] nlme_3.1-157 mime_0.12 formatR_1.14
## [85] compiler_4.2.0 plotly_4.10.1 png_0.1-8
## [88] spatstat.utils_3.0-1 tibble_3.1.8 bslib_0.4.2
## [91] stringi_1.7.12 highr_0.10 desc_1.4.2
## [94] lattice_0.20-45 Matrix_1.5-3 vctrs_0.5.2
## [97] pillar_1.8.1 lifecycle_1.0.3 spatstat.geom_3.0-6
## [100] lmtest_0.9-40 jquerylib_0.1.4 RcppAnnoy_0.0.20
## [103] data.table_1.14.8 cowplot_1.1.1 irlba_2.3.5.1
## [106] httpuv_1.6.9 patchwork_1.1.2 R6_2.5.1
## [109] promises_1.2.0.1 KernSmooth_2.23-20 gridExtra_2.3
## [112] parallelly_1.34.0 codetools_0.2-18 MASS_7.3-56
## [115] rprojroot_2.0.3 withr_2.5.0 sctransform_0.3.5
## [118] parallel_4.2.0 grid_4.2.0 tidyr_1.3.0
## [121] rmarkdown_2.20 Rtsne_0.16 spatstat.explore_3.0-6
## [124] shiny_1.7.4