We provide a series of vignettes, tutorials, and analysis walkthroughs to help users get started with Seurat. You can also check out our Reference page which contains a full list of functions available to users.
Introductory Vignettes
For new users of Seurat, we suggest starting with a guided walk through of a dataset of 2,700 Peripheral Blood Mononuclear Cells (PBMCs) made publicly available by 10X Genomics. This tutorial implements the major components of a standard unsupervised clustering workflow including QC and data filtration, calculation of high-variance genes, dimensional reduction, graph-based clustering, and the identification of cluster markers.
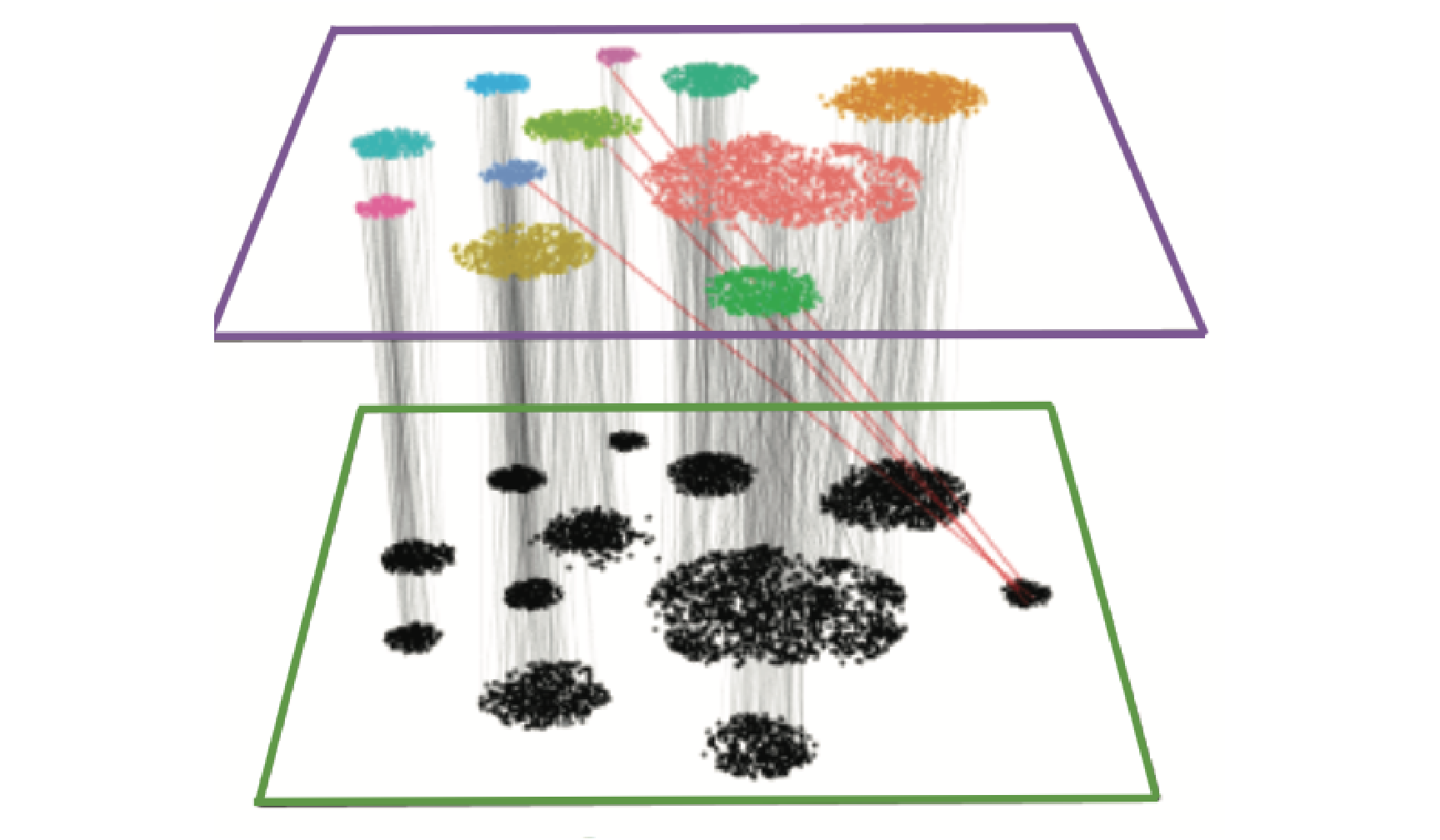
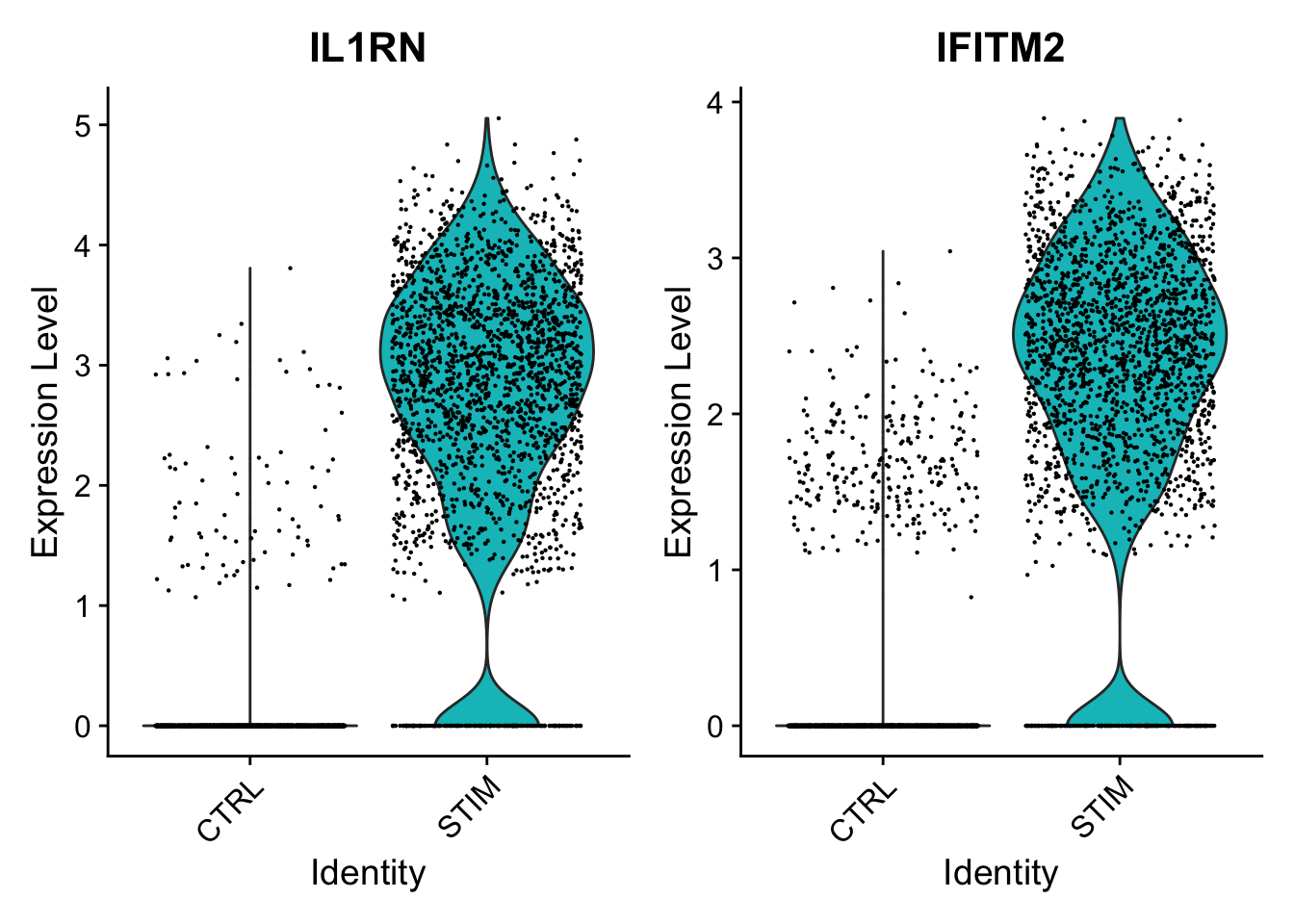
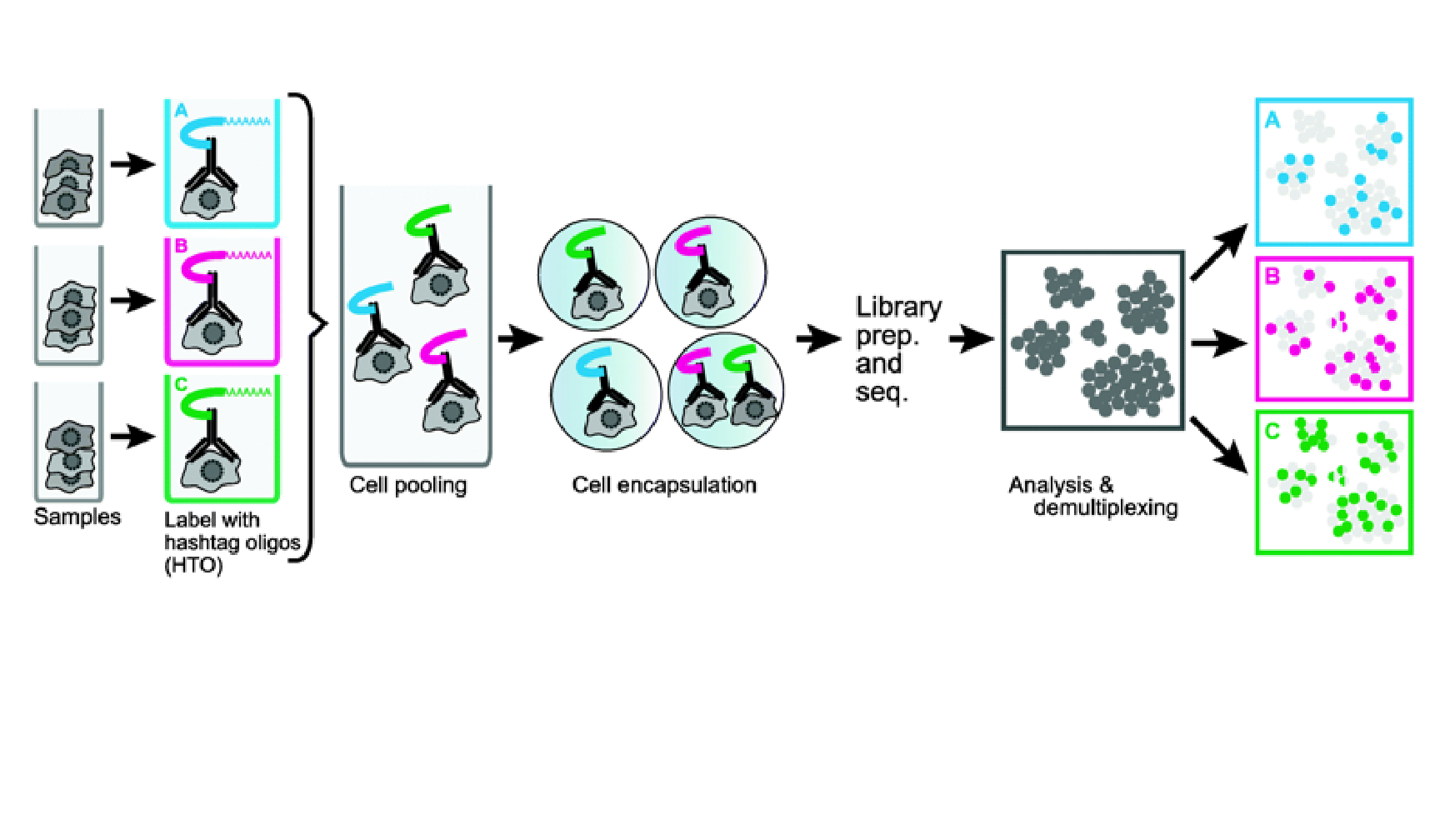
We provide additional introductory vignettes for users who are interested in analyzing multimodal single-cell datasets (e.g. from CITE-seq, or the 10x multiome kit), or spatial datasets (e.g. 10x Visium or Vizgen MERFISH).
|
|
|
|

|

|

|
|
A basic overview of Seurat that includes an introduction to common analytical workflows.
|
An introduction to working with multi-modal datasets in Seurat.
|
Learn to explore spatially-resolved transcriptomic data with examples from 10x Visium and Slide-seq v2.
|
|
GO
|
GO
|
GO
|
|
|

|
|
Learn to explore spatially-resolved data from multiplexed imaging technologies, including MERFISH, Xenium, CosMx SMI, and CODEX.
|
|
GO
|
SeuratWrappers
In order to facilitate the use of community tools with Seurat, we provide the Seurat Wrappers package, which contains code to run other analysis tools on Seurat objects. For the initial release, we provide wrappers for a few packages in the table below but would encourage other package developers interested in interfacing with Seurat to check out our contributor guide here.
|
Package
|
Vignette
|
Reference
|
Source
|
|
alevin
|
Import alevin counts into Seurat
|
Srivastava et. al., Genome Biology 2019
|
https://github.com/k3yavi/alevin-Rtools
|
|
ALRA
|
Zero-preserving imputation with ALRA
|
Linderman et al, bioRxiv 2018
|
https://github.com/KlugerLab/ALRA
|
|
CoGAPS
|
Running CoGAPS on Seurat Objects
|
Stein-O’Brien et al, Cell Systems 2019
|
https://www.bioconductor.org/packages/release/bioc/html/CoGAPS.html
|
|
Conos
|
Integration of datasets using Conos
|
Barkas et al, Nature Methods 2019
|
https://github.com/hms-dbmi/conos
|
|
fastMNN
|
Running fastMNN on Seurat Objects
|
Haghverdi et al, Nature Biotechnology 2018
|
https://bioconductor.org/packages/release/bioc/html/scran.html
|
|
glmpca
|
Running GLM-PCA on a Seurat Object
|
Townes et al, Genome Biology 2019
|
https://github.com/willtownes/glmpca
|
|
Harmony
|
Integration of datasets using Harmony
|
Korsunsky et al, Nature Methods 2019
|
https://github.com/immunogenomics/harmony
|
|
LIGER
|
Integrating Seurat objects using LIGER
|
Welch et al, Cell 2019
|
https://github.com/MacoskoLab/liger
|
|
Monocle3
|
Calculating Trajectories with Monocle 3 and Seurat
|
Cao et al, Nature 2019
|
https://cole-trapnell-lab.github.io/monocle3
|
|
Nebulosa
|
Visualization of gene expression with Nebulosa
|
Jose Alquicira-Hernandez and Joseph E. Powell, Under Review
|
https://github.com/powellgenomicslab/Nebulosa
|
|
schex
|
Using schex with Seurat
|
Freytag, R package 2019
|
https://github.com/SaskiaFreytag/schex
|
|
scVelo
|
Estimating RNA Velocity using Seurat and scVelo
|
Bergen et al, bioRxiv 2019
|
https://scvelo.readthedocs.io/
|
|
Velocity
|
Estimating RNA Velocity using Seurat
|
La Manno et al, Nature 2018
|
https://velocyto.org
|
|
CIPR
|
Using CIPR with human PBMC data
|
Ekiz et. al., BMC Bioinformatics 2020
|
https://github.com/atakanekiz/CIPR-Package
|
|
miQC
|
Running miQC on Seurat objects
|
Hippen et. al., bioRxiv 2021
|
https://github.com/greenelab/miQC
|
|
tricycle
|
Running estimate_cycle_position from tricycle on Seurat Objects
|
Zheng et. al., bioRxiv 2021
|
https://www.bioconductor.org/packages/release/bioc/html/tricycle.html
|